
Bio-decontamination is a critical element of manufacturing and other cleanroom activities. But this process is not complete unless the efforts are fully validated.
Validating and understanding bio-decontamination processes is vital, but requirements have been elevated by the implementation of revised Annex 1 guidelines this year. In particular, changes surrounding bio-decontamination validation mean manufacturers of sterile products must ensure they comply with more detailed requirements than previously.
Further information on Protak Scientific is available at www.protakscientific.com and you can get in touch with us at contact@protakscientific.com
This presents a number of challenges, but with the right technology, in particular, the use of Enzyme Indicators, equipment suppliers and product manufacturers can step up and deliver a consistent, highly effective solution for bio-decontamination validation throughout the whole manufacturing lifecycle.
What is bio-decontamination?
Bio-decontamination refers to essential processes used to destroy, eliminate or reduce all forms of microbial life from surfaces in the inanimate environment. This includes vegetative bacteria, bacterial spores, fungi, fungal spores, and viruses. For critical equipment and/or processes that cannot be terminally sterilised via steam sterilisation or ethylene oxide, this is typically achieved through the automated use of gaseous hydrogen peroxide (H2O2) injected into the environment.
An effective bio-decontamination process should have defined cycle parameters that are robust, repeatable and provide highly controlled conditions within the enclosure. This must deliver a consistent supply of H2O2 concentration, capable of rendering the internal surfaces and any load items decontaminated to a suitable Sterility Assurance level (SAL). The SAL dictates the probability of a viable microorganism surviving after the bio-decontamination process and is typically set within the range of 104 to 106. The time to remove and evacuate the H2O2 from the enclosure space is also of utmost importance.
What most may not realise, however, is just how many potential variables there are in the process that can impact the efficacy of the bio-decontamination process. This can mean that even if the same automated starting cycle process is followed, the results may not achieve the required SAL, and without investigation or visibility as to the factors behind this, it makes it hard to determine the cause of a failure or replicate the conditions.
For example, the temperature and humidity of the enclosure can have a major impact on the efficacy of the H2O2 decontamination process. If this is not controlled carefully, even a small enclosure can have variation that impacts H2O2 concentration and allows failures to occur. Similarly, load configuration and construction materials can also impact the procedure due to materials being permeable to H2O2 and causing H2O2 concentration variation.
Gloves/gauntlets used within isolators also represent a challenge as they are a permeable material, as well as being irregular shape and at risk from occlusion from the H2O2 distribution. This is a hot topic that has been worked specifically into the Annex 1 section 4.22 requirements, which state: “Gloves should be appropriately extended with fingers separated to ensure contact with the agent.”
This is why an effective validation solution is so important to the process. Without this, manufacturers run the risk of uncontrolled and ineffective bio-decontamination processes that can have a serious impact on the sterility assurance of the product or process that is being employed.
How are bio-decontamination processes validated?
The bio-decontamination validation phase of any project can become a lengthy process when the design, use and critical performance parameters (CPP) have not been considered during the design phase. As such, it is important to fully understand how validation can be integrated throughout the lifecycle.
This starts at the initial engineering stage with drafting comprehensive User Requirement Specifications to set out the key operating conditions and ranges right from the start. This should include key questions such as how long cycles need to be in operational use and how many cycles will be needed.
Specifying the right conditions to ensure repeatability and the maximum loading of materials that are needed are also essential elements of user requirements that must be determined during initial engineering.
Clear answers to all these are vital to avoid failures within the late stages and ensure the enclosure and its bio-decontamination is fit for purpose. Testing at this stage must replicate the real-world conditions and parameters as closely as possible to avoid skewing the results.
After User Requirement Specifications (URS) have been established and tested, there are several steps that must be followed. For instance, once into the engineering/cycle development phase, the above questions should be put into practice and robustly challenged to ensure that the boundaries of operation have been confirmed as effective for the intended use.
Performance Qualification (PQ) validation is also critical to show the process can be repeated and give consistent SAL results. Bio-decontamination validation is not complete until these results have been verified.
What are the challenges of validation?
Traditionally, this involves the use of biological indicators (BIs) and chemical indicators (CIs), which are positioned around the enclosure in locations that represent a challenge to the H2O2 distribution, before being incubated in a growth media for seven days. If growth appears at any point in this timeframe, shown by turbidity in the media, this indicates a failure.
This is a tried-and-tested process that has been used in some form for decades. However, this does not mean that it is straightforward or easy to perfect. The use of conventional bio-decontamination validation methods comes with a number of challenges that must be carefully addressed in order to obtain clear, consistent results.
Validation may also fail if the system is not optimised mechanically - from the materials used in chambers to equipment such as gloves. The selection of the right location within the enclosure is another challenge. Traditionally, there has been a lot of guesswork involved in this process and there is varied guidance on how many indicators should be used in a given space or where they should be located.
Finally, indicators like BIs give a binary pass/fail answer that does not provide enough data to refine processes, determine how close to success the procedure was, or to fully understand why failures have occurred.
How do Enzyme Indicators bring value?
Many of these challenges can be overcome with the use of Enzyme Indicators (EIs). These offer a fast, reliable alternative to conventional methods that can be applied to any process where gaseous H2O2 bio-decontamination is used.

Picture of packet and test tubes
The main benefits of this approach are greatly reduced validation execution times and quantifiable efficacy data, which in turn lead to significant cost savings. Whereas other methods need seven days to confirm a result, EIs provide answers in 60 seconds. This isn't just faster - it eliminates the risk of a failure, where consequences could include product losses and ultimately increased manufacturing costs.
Under traditional approaches, it can be easy to miss problem areas. EIs eliminate this issue and take all of the guesswork out of validation processes. The EI technology lets users map multiple locations to learn exactly where the most problematic areas are, resulting in a full distribution model. This in turn allows the possibility of challenge location reduction during the next phases of the validation lifecycle with data-driven assessment.
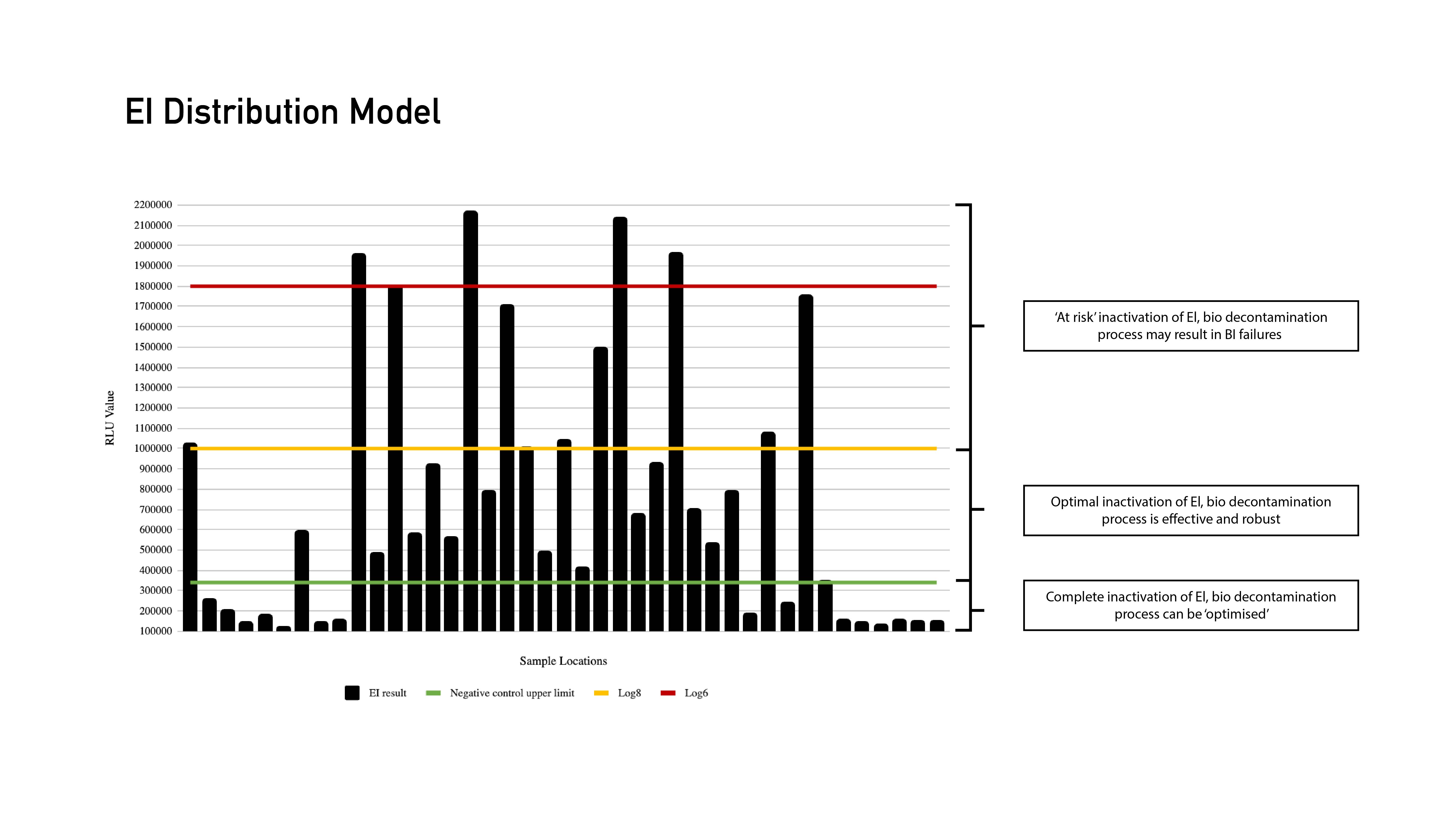
EI Distribution Model
Adding EIs early sets up good initial cycles, and the better data this provides funnels down to the rest of the validation process, making every stage faster. In one real-world example for a major pharmaceutical company, this reduced cycle redevelopment from 17 cycles across 12 weeks to just six cycles in three weeks.
Protak Scientific's EI approach is not just a technology solution - they also provide the essential consultancy and strategy expertise to minimise the risk of a costly bio-decontamination validation failure. By treating validation as a holistic process and using their wealth of knowledge, they can offer best in class global technology solutions for bio-decontamination.
This can range from managing the development cycle or advising on Annex 1 issues such as the use and most effective decontamination assessment of gloves. Their capability is to fill in the gap between user requirements specifications and the delivered final process.
Ultimately, relying on legacy technology means leaving validation to chance and hoping that results will meet the required SAL. But hope is not a strategy for bio-decontamination. The use of EIs eliminates this risk and offers a bio-decontamination validation solution that is an integral part of every step of the development process and greatly cuts both time and cost for users. This results not only in a successful validation, but also ensures compliance with the Annex 1 requirement that the process is understood and validated.
Image of Equipment with the product and Aphena PC
Further information on Protak Scientific is available at www.protakscientific.com and you can get in touch with us at contact@protakscientific.com






