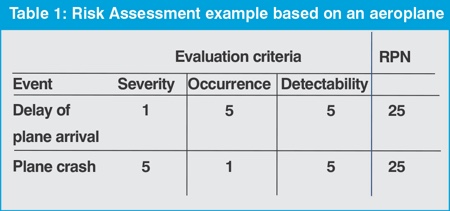
Over the past decade, methods of risk analysis have been actively propagated for pharmaceutical manufacturing, and companies are strongly encouraged to apply these methods. However, do such risk analyses have any real value?
Broadly speaking, a risk is the possibility that a dangerous or unwanted event may occur. It could be financial, traffic-related or to do with equipment faults, safety (e.g. with nuclear power stations). People spend time every day estimating or analysing risks to protect themselves against unwanted events. The purpose of risk analysis is to understand the ‘Reasons–Consequences’ chain with a view to finding the best protection. But any new method should be supported by demonstrations of its effectiveness and practicality. The medicinal products sector, however, has a special feature in that no risk is acceptable.






