
In medical device production low bioburden levels are key. Bioburden control and how it can affect product sterilisation was the topic of a recent GMP seminar by sterilisation service provider Isotron. Susan Birks was there to report.
The seminar organised by Isotron targeted a range of cleanroom users, from medical device producers through to contract packers and component suppliers. The topic was good manufacturing practice (GMP) and the presentations covered the basic principles of cleanroom operation, the regulations, tests for bioburden and endotoxins, reasons and requirements for environmental monitoring, how to handle “out of spec” results and finally some essential dos and don’ts from guest speakers with long experience in cleanroom use.
Cleanrooms are constantly under siege from several enemies labelled broadly as contamination. In most instances, the contaminants are invisible to the naked eye, which makes monitoring their presence a difficult but crucial task. The battle against this enemy is also subject to constantly moving goalposts, as the regulatory bodies are moving away from prescriptive regulatory measures to a risk-based approach, with the burden of risk analysis and proof falling on the shoulders of the cleanroom operators.
Microbiology expert Peter Rose, managing director of cleanroom consultancy High Edge Consulting, gave an overview of the basic principles and compliance requirements for medical device production. The main mandated requirements, he said, include that expressed in ISO 13485:2003 paragraph 6.4 on work environment: The organisation shall determine and manage the work environment needed to achieve conformity to product requirements; and that of the US FDA Quality System Regulation 21 CFR 820.70(c) on Environmental control, which states: “…the manufacturer shall establish and maintain procedures to adequately control these environmental conditions…”
However, when commissioning a cleanroom there are no hard and fast rules to follow. Rose said: “There are norms and expectations, but as every product is different, companies need to do their own risk analysis on their products and processes to establish that the end product is risk-free to patients.”
As a result, terminally sterilised medical devices do not have specific cleanroom classification requirements. Most, however, are between Class 7 and 8. (Aseptic products do have specified requirements, covered in EN (ISO) 13824 and EN 14160 and EU Good Manufacturing Practices and ISO 14644-1).
When deciding which cleanroom classification to build, it is worth considering that the cost difference between levels 7 and 8 is not that great, said Rose, so many companies opt for a Class 7 as an added safety factor.
The concept that medical device manufacturers need to keep in mind is that “validation of device sterilisation is dependent on the device’s pre-sterilisation bioburden,” Rose stressed, and this is true for all sterilisation technologies, but particularly for irradiation. This means that if bioburden levels rise in the cleanroom, then the “validation of sterilisation” previously carried out is no longer valid. To avoid this, “companies need proof of bioburden control, which means they need routine pre-sterilisation biodata, i.e. product bioburden and room environmental data from critical control points.
“A common misconception is that cleanrooms clean things, when in fact cleanrooms are only as clean as the products that are introduced into them,” he said.
To maintain cleanroom bioburden levels, all sources of contamination (people, products, packaging and cleaning materials) need to be controlled (see Table 1).
| Table 1: Types of contamination | |||
It is estimated that some 80% of contamination (skin, hair and microbes) comes from people entering the cleanrooms. The more active the people are, the more contamination they shed. To limit this, operatives must be trained to move slowly, not to talk to other operatives across the room, and not to wear cosmetics (including nail varnish and perfume). Ideally, they should not be smokers or have respiratory diseases or skin conditions.
Apart from all the necessary training in hand washing and appropriate gowning, operatives must be in a clean state when arriving for work. Experience has shown that activities carried out prior to entering the workplace can affect levels of bioburden – wallpapering at home, for example, was discovered to be the cause of a sudden increase in spores in one cleanroom.
It is also important for medical device producers to carry out their bioburden tests for validation of sterilisation just prior to the product being sterilised. “If product is being left around on shelves, companies need to redo the bioburden tests, as bioburden may have changed during storage,” said Rose.
The consequences of losing the battle with the invisible enemy will be seen only once the product has been used by, or is inserted into, the patient, and can include infection, allergic reaction, toxic reaction or even physical damage from something as seemingly innocuous as fibres which, if left on devices used intravenously, can strip out valves in blood vessels.
Endotoxin
One of the invisible enemies frequently overlooked is endotoxins – fragments of dead Gram negative bacteria that are toxic to the human body and that can cause fever or fatal reactions. Rose emphasised the need for companies to determine whether their devices carry endotoxins.
Limulus Amebocyte Lysate (LAL) is the test reagent. It reacts specifically with bacterial endotoxin, highlighting its presence. For a product to carry any kind of claim, such as “endotoxin free” or “non-pyrogenic”, tests must be carried out to prove this.
A frequent source of endotoxins and bioburden is water, which can carry Gram negative bacteria. It is therefore important to examine process water usage. Endotoxin can come from water further back down the supply chain – for example from extrusion, grinding, milling or cleaning processes.
The fact that endotoxins are produced by the death of bacteria means, paradoxically, that sterilisation can sometimes liberate more endotoxins. Prevention is the preferred method of controlling them, particularly since the removal treatments require high temperature and strong chemicals, which are not suitable for most plastic materials.
Rose said the expectation is that when companies test devices for bioburden, the associated risk assessment should also include whether it is necessary to test for endotoxins. But as a rule of thumb: “If water is present, I would test,” he said.
Regulations
Stewart Brain, microbiology team leader, BSI Healthcare, gave further clarification on the regulations for medical devices. In particular, he highlighted the Medical Devices Directive (MDD) 93/42/EEC Essential Requirement 8.5: ‘Devices intended to be sterilised must be manufactured in appropriately controlled (e.g. environmentally) conditions’.
He also outlined key parts from the international standard ISO 14644 1–9 that relate to cleanrooms. In particular, he noted the importance of Part 1, which gives classification by particle size (airborne particle concentration limits), and Part 2, which gives details of test frequencies for airborne particle concentration limits, airflow volume or airflow velocity, air pressure differential.
Part 2 also outlines that the monitoring regime should be based on a risk assessment and gives requirements for requalification.
Key issues that Brain identified from the MDD are:
- The work environment must be periodically monitored to verify the effectiveness of the control measures;
- The monitoring regime should include microbiological testing (e.g. settle plates, air sampling and surface sampling and non-viable testing (e.g. particulates, differential pressure).
Brain also looked at some of the variations and additional requirements of other countries such as Australia and Japan.
Finally, he highlighted the importance of training, not only for frequent users and full time operatives, but also for contract maintenance workers, or quality personnel who may enter the cleanroom infrequently to collect samples for validation.
Su Finlay-Woods, lab manager at Isotron’s South Marston Laboratory, looked at the concepts and different grades of cleanroom, the different airflow patterns and how these can affect operational practices. “As most airflows direct the invisible contamination downwards and away from workstations to the floor, it is important to remember not to store anything on the floor area, as that will be the dirtiest area in the room,” she said. It is also important not to place equipment or products where they can block vents and thus restrict the effectiveness of the airflow.
Finlay-Woods also gave an interesting analogy as to why it is important to be properly gowned and not wearing make-up when in cleanrooms. Microbes will tend to float around on dust particles, rather than on their own, and whereas small particles of dust form a life-raft for transporting microbes, skin cells stuck together with make-up form cruise liners, she said.
In addition to outlining methods of cleaning and monitoring, Finlay–Woods also emphasised the importance of documenting these procedures in SOPs.
Gary Ward, lab manager at Isotron’s Thorne laboratory, looked in more depth at environmental monitoring procedures and, once again, highlighted the fact that if one cannot control the bioburden, one cannot control the sterility assurance level (SAL) of products.
In terms of standards and guidelines, environmental monitoring is covered by:
- BS EN ISO 14698-1 Cleanrooms and associated environments – Biocontamination parts 1 & 2;
- ISO 14644 – Cleanrooms and associated controlled environments Parts 1–7;
- US Federal standard 209E Airborne Particulate Cleanliness Classes in Cleanrooms and Clean Zones;
- EU cGMP Annexe 1; and
- USP Chapter 116, Microbiological evaluation of cleanrooms.
Ward looked at the main microbiological contaminants of interest to the medical device and pharma sectors – bacteria and fungi – and at how to monitor levels using active air sampling, passive sampling, settle plates and sampling of surfaces. He also reviewed the types of growth media and incubation conditions.
In addition to giving advice on designing a monitoring programme, Ward outlined how to go about room qualification, collecting data and, more important, tending data. “The purpose of environmental monitoring is to correct problems before product is placed at risk,” he said. “Therefore it is necessary to trend data on a regular basis”.
Out of spec results
Rose then advised on handling “out of specification” results. He said that bioburden limits are set for two reasons:
1) As acceptable and safe levels, and
2) As indicators of the normal range.
Two types of limits need to be set: an Alert limit and an Action limit. SOPs should give guidance on routine responses to breaches of both of these. Outlining what action has to be taken can help operators through low level out of spec incidences without the need for unwarranted product recalls, he said.
When setting the limits, it is important that they are not only safe and acceptable for the product concerned but are also realistic. It may be worth taking time to decide the right limits and review them a year later as seasonal variations can affect results.
“If out of spec results do occur, don’t panic. Contain the problem first, then think logically about what action to take,” he said.
Rose advised companies to resist the temptation simply to do a retest without a good reason. “If you do so, you will just end up with two different results. It is better to do several retests, and if you get three or four out of five that fail, then you know you have a problem,” he said.
It is helpful to record the activities going on in the room when the settle plates are put out, as any out of spec results can then be investigated in relation to what was happening in the room at the time.
When seeking the source of contamination, look for changes in the types of organisms being found, as this could help. It is also worth cross-referencing with environmental monitoring results, he said. Questions to pose are:
- Has the water source changed?
- Are there new neighbours to the building?
- Has any maintenance been carried out?
- Has the cleaning regime changed?
- Have the raw materials changed?
- Has the shift pattern changed?
- Have new sub contractors been employed?
- Have suppliers changed?
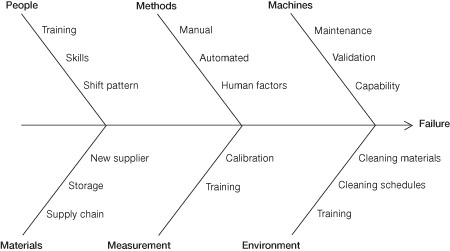
Ishikawa diagram principles (fishbone diagram)
Something that many companies forget to do is a product impact assessment. Does the out of spec result affect the product? It may be that something has pushed the product outside the normal range of the specification but the product may still be safe. Carry out endotoxin tests, suggests Rose. If the tests show there is no impact on the product, there is no need to reject it.
In the final sessions, two experienced cleanroom users – Russ Bird, managing director of cleanroom consumables supplier Basan, and Jon Charters, QA/RA manager, Stanmore Implants – offered their top dos and don’ts when building or operating a cleanroom.
Bird stressed the importance of good design before building. Things to consider were:
- What cleaning methods will be used, by who and how?
- How will cleaning materials be stored and brought into the cleanroom?
- Where will cleaning agents be prepared?
- Where and how will used water and agents be disposed of ?
- What is the changing room access and frequency of cleaning (as the dirtiest area)?
- Is the site suitable? (i.e. not near a doorway or lorry unloading park)
Charters explained how the supplier of bespoke medical implants, Stanmore Implants, employs the services of Isotron to help with its routine monitoring programme. For example, it uses actual components as samples that it puts through the cleanroom process and then sends them to Isotron for bioburden analysis. It also takes monthly water samples, monthly settle and contact plate counts (using plates supplied by Isotron) and quarterly particle counts from predefined monitoring locations set out with a defined protocol.
The company receives a report back from Isotron detailing the CFU levels for the various samples. If the limits have been exceeded the company is also informed by phone or e-mail prior to receiving the report so action can be taken. The results are also analysed on a monthly basis to look for trends. In this way, Stanmore keeps its cleanroom under control, is compliant with regulations and is confident that it is minimising patient risk.
The day served to provide the basics for newcomers and a reminder for old hands of common mistakes in cleanroom operation. More importantly, it provided a reminder that people are the main perpetrators of contamination and training is key for all those entering a contamination controlled environment.

Featured companies

- Companies:
- BSI British Standards
- Stanmore Implants
- Avantor Sciences

