Any analytical method applied to a pharmaceutical product under current Good Manufacturing Practices (cGMPs) requires validation. The methods used to produce data supporting the production of pharmaceuticals or regulatory filings (with FDA for example) need to be validated prior to use.
Validated analytical methods support a drug’s characterisation, quality control and manufacturing batch records. This analytical test method validation provides a documented process demonstrating that the test procedure is suitable for its intended purpose, provides evidence of the method’s performance and ensures quality and reliability of results.
Data quality
A test method validation can be seen as one of the components of an overall process of generating consistent and reliable quality data (Figure 1).1 The first key component that forms the base for generating the quality data is Analytical Instrument Qualification, which is a collection of documented evidence that the instruments used in the method perform suitably for their intended purpose.
It is critical to qualify any equipment, software or database by employing Installation, Operational and Performance Qualifications (IQ, OQ and PQ) prior to method validation.
Using a validated procedure with qualified instruments gives confidence that the procedure will generate valid data of acceptable quality. System Suitability Tests verify the performance of the test in accordance with the criteria set in the procedure and ensure acceptance of this performance at the time of the test; they are based on the principle that the equipment, electronics, analytical operations and samples to be analysed form an integral test system.
Together with Quality Control Check samples, which provide an ongoing assurance of the test’s performance, system suitability tests ensure that the validated method will work under the specific conditions. Verification, preparatory testing or product specific validation can all be classified as a part of system suitability testing.
Guidelines and requirements
Even though method validation is required for all pharma test methods, the validation process itself can vary greatly depending on the regulatory bodies governing drug development, production and the purpose of the method. As government regulations do not specify in detail the scientific approach to method validation, pharmaceutical companies and contract analytical testing laboratories rely on guidelines to meet the expectations of compliance and scientific soundness of the method in use.
Regulatory and government bodies such as ICH or FDA provide sets of guidelines that, while being non-binding, conform to the regulatory requirements and industry standards of safe pharmaceutical product development and provide a set of official references regarding method validation. Examples of available guidance documents are
- Validation of Analytical Procedures: Text and Methodology Q2(R1), ICH 2005
- Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics, FDA 2015
- Guidance for Industry: Bioanalytical Method Validation (Draft Guidance), FDA 2013
- <1225> Validation of Compendial Procedures, USP 39
- <1226> Verification of Compendial Procedures
- <1058> Analytical Instrument Qualification.
Guidance documents provided by regulatory bodies describe a number of characteristics or performance elements that are to be defined during the process of method validation. The specific combination of characteristics to be evaluated in the process of validation depends on the nature of the particular test procedure; assay validation parameters can vary greatly between methods, depending on the purpose of the assay or the compound of interest.
Validation will, therefore, document through specific laboratory examinations that the method’s performance characteristics are reliable and suitable for the intended use. The criteria used to validate the acceptability of analytical data needs to be defined early in the process alongside the scope of the method.
According to Validation of Analytical Procedures: Text and Methodology Q2(R1),2 these characteristics are
Accuracy: the closeness of test results obtained by the method to the true value, which is either accepted as a conventional true value or reference value. Frequently the evaluation of accuracy will involve the use of reference standards or calculation of the percentage of recovery of a known added amount of analyte in a sample. As the analytical method is expected to be accurate across its range, the accuracy should be assessed throughout the method’s expected range of values.
Precision: the closeness of agreement between the series of measurements of multiple sampling of the same homogenous sample under strictly defined experimental conditions. It should represent the method’s repeatability and reproducibility. As in the case of accuracy determination, it has to be assessed throughout the expected range of measurements.
Specificity: the ability to assess categorically the analyte in the presence of components that may be expected to be present, such as impurities, degradation products and matrix; a highly specific method will detect the article of interest, even at very low quantities, in the presence of high quantities of other similar but not identical articles. Again, the determination of a procedure’s specificity has to be performed across the expected range of values and under strictly defined conditions.
Detection limit of an analytical method is the lowest amount of analyte in a sample that can be detected but not necessarily quantitated as an exact value under defined experimental conditions. It constitutes the lowest amount of tested material contained within the analysed sample that produces a signal above the noise.
Quantitation limit of an analytical procedure is the lowest amount of analyte in a sample that can be quantitatively determined with suitable precision and accuracy under defined experimental conditions. The established quantitation limit will define the lowest point of the range of the method in question.
Linearity of an analytical method represents its ability, within a given range, to obtain test results that are directly, or by well defined mathematical transformation, proportional to the concentration of the analyte in the sample. If linearity cannot be attained, a non-linear model can be used. Any quantitative method must demonstrate a defined quantitative relationship between the assay measurement and the quantity of material of interest in the tested sample. Range of an analytical procedure is the interval between the upper and lower levels of analyte in the sample (including these levels) for which it has been demonstrated that the analytical procedure has suitable precision, accuracy and linearity.
Robustness of an analytical method measures its capacity to remain unaffected by small, but deliberate variations in method parameters and provides an indication of its reliability during normal usage.
Types of method validation
To demonstrate the validity of the method’s performance, depending on the methodology used and the purpose of the assay, the analytical procedure needs to undergo one of the various types and levels of validation. In general, method validations fall into three categories: full validation, partial validation and cross-validation.
Full validation is essential for newly developed and implemented methods or when changes applied to the existing method have an impact on critical components or the scope of the procedure. Partial validation applies to already validated methods that undergo further modifications. Changes in detection system, sample processing, quantitation range, composition of matrix and many others warrant partial validation.
The choice of performance characteristics assessed in partial validation should be based on possible effects that the new changes could have on the method’s execution. Verification process can be classified as partial validation. Cross-validation compares validation parameters when two or more methods are used to generate data within the same study or across different studies. It is performed to determine the inter-laboratory reliability or equivalency of different analytical techniques. Moreover, some changes applied to previously validated methods can merit further revalidation of the analytical method that, depending on the extent of changes, can substantiate either a full or partial validation.
Method verification
Importantly, not all methods have to be fully validated before they are first used in laboratories. Compendial methods, those published in monographs and general chapters of Pharmacopoeias (USP and Ph. Eur.) ‘have been validated in accordance with accepted scientific practice and current recommendations of analytical validation. Unless otherwise stated in the monograph or general chapter, validation of the test methods by the analyst is not required.’3
Nevertheless, when applying a pharmacopoeial method, the user is required to establish a documented evidence of suitability of the method under actual conditions of use; that is, verify that the conditions under which a validated method is applied to test a particular product are suitable for the method in question. Subsequently, verification of a compendial test procedure aims to determine whether, under the actual conditions of use and for the specified substance or drug product matrix, the procedure in question can be used for its intended purpose.4
Verifying a method’s suitability under actual conditions of use does not require a complete revalidation of a compendial method. Instead, it focuses on the assessment of some of the analytical characteristics evaluated during the validation; that is, the performance elements most representative of the critical aspects of the method. The extent of the verification process depends on many factors: type of method and equipment used, type of tested product, procedure steps or even the user’s experience and training. Nevertheless, ultimately the user (pharmaceutical companies, contract analytical testing labs) is responsible for selecting which of the performance characteristics assessed during the validation process should be included in the verification protocol.
Lifecycle of an analytical method
The lifecycle of an analytical method starts when a pharmaceutical company or a contract analytical testing laboratory recognises a requirement for a new analytical method (Figure 2). They will then either identify an existing/compendial procedure suitable for the particular need or proceed to develop a completely new method. Following the development stage, a validation (or verification) plan will be prepared and the method validation experiments will be performed.
The process of validation should follow a validation protocol which must clearly define the application purpose and scope of the method, performance characteristics with acceptance criteria, validation experiments, standards and reagents. Once the analytical method validation confirms the method’s suitability for its intended purpose, the standard operating procedures (SOPs) for the routine execution of the method need to be developed and approved.
As the analytical method should be continually monitored for its fitness for purpose throughout its lifecycle, the criteria for revalidation and type/frequency of system suitability tests and QC checks should be defined. Following the successful submission of the validation report, the analytical procedure can be used for routine analysis. If changes to the method occur, the evaluation of their effect on the procedure’s suitability for its intended use is essential. If changes applied to the analytical method are covered by current validation, no further validation is necessary. Otherwise, any changes falling beyond the scope of existing validation will result in either revalidation or, sometimes, method redevelopment and new validation (Figure 2).
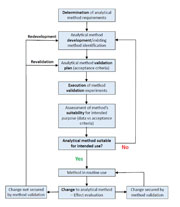
Figure 2: Analytical method development and validation: the lifecycle of a method
Data generated via the analytical method application in the pharmaceutical industry is crucial for many of the critical decisions that ultimately ensure the health and safety of consumers. Consequently, it comes as no surprise that validation of an analytical method is a highly scrutinised process – it is of paramount importance that the user is confident in the integrity of this data and that the method is fit for purpose. For the highest quality of the method’s performance, a lifecycle approach to method validation is applied as it reflects the integration of the method with the operating environment and allows continuous monitoring and improvement. Last but not least, the level of training and scientific expertise of staff planning and performing validation should not be underestimated as they are an integral part of the strong foundations of high quality method performance.
References
1. United States Pharmacopoeia 39 - NF 34 (2016), <1058> Analytical Instrument Qualification.
2. International Conference on Harmonization (ICH) Quality Guidelines Q2(R1) (2005), Validation of Analytical Procedures: Text and Methodology.
3. General Notices, European Pharmacopoeia 8th Edition (2016).
4. United States Pharmacopoeia 39 - NF 34 (2016), <1226> Verification of Compendial Procedure.
