The bacterial aspect of molecular analysis is particularly interesting as is now well known from the literature that cannabis and other flowering plants present residual “bacterial 16S-like” sequences in both their chloroplast and mitochondrial genomes.
Addressing these findings, PathogenDx has recently been awarded a pair of US Patents #10,501, 814 (December 10, 2019) and #10,513,745 (December 24, 2019), Hogan Michael Edward, et al. The invention describes a method of using generic primers for PCR amplification of microbial samples, both bacterial and fungal, followed by microarray analysis of the resulting PCR products.
This is a technology patent and as such we have described in it a number of primer-probe combinations, some of which are used in our current products and others which are not.
The general technology platform described in the patent has also led to the design of probes and primers that are similar to those in the patent, but not yet included as a Continuation in Part of the patent and thus remain, for now, as trade secrets. Because of significant interest in this patent, we have chosen to describe in more detail, in this publication, the core attributes of the patented technology and to show an example of a specific primer-pair design from the patent, which serves as a good demonstration of the specificity of the technology.
Focus on 16S DNA testing
A major focus and generally useful aspect of the invention described in Patents 10,501,814 and 10,513,745 is the analysis of bacterial samples via testing of the 16S DNA in them, especially bacterial samples that have been recovered from plant matter and analysed without enrichment culture. Under those culture-free conditions, there is a possibility that residual plant tissue may be recovered along with the contaminating bacterial cells of interest.
Consequently, a major focus of Patents 10,501,814 and 10,513,745 is a methodology to selectively amplify the “true” bacterial 16S DNA in these recovered samples, under conditions where “bacterial16S-like” DNA within plant chloroplast and mitochondria is not PCR amplified and consequently, remains invisible during microarray hybridisation analysis thereafter.
A number of scientists in the cannabis community have shown interest in better understanding the process described in PathogenDx Patents 10,501,814 and 10,513,745, with a special focus on such 16S DNA discrimination. The process is relatively simple and is based on the general structure of the PathogenDx microarray assay, as described in Patents 10,501,814 and 10,513,745

Mike Hogan
Tandem PCR + Microarray assay overview
Sample preparation and analysis in these assays is relatively simple and follows the same overall process:
- Harvest of a microbial cell pellet, usually from a plant wash by centrifugation or filtration
- Lysis of the resulting microbial pellet to release the DNA content of the cells.
- PCR amplification of the raw pellet lysate “loci enhancement” to generate a “Primary Amplicon”. The loci enhancement reaction amplifies a ≈500bp region of bacterial 16S DNA, but not the “16S-like” sequences.
- PCR amplification of the Primary Amplicon “labelling PCR” to generate a Secondary Amplicon. The labelling PCR reaction is nested within the 500bp domain to generate a ≈225bp sub-region of 16S DNA.
The orientation of those two PCR reactions on the underlying bacterial 16S DNA gene is shown in Figure 1, which is a summary of the data described in Patents 10,501,814 and 10,513,745.
- Loci enhancement is driven by PCR primers identified as: a. Seq 1 (forward PCR primer) and Seq 2 (reverse PCR primer).
- Labelling PCR is driven by PCR primers identified as: a. Seq 19 (forward PCR primer) and Seq 20 (reverse PCR primer).
The PCR primer pair that drives labelling PCR (Seq 19, 20, red boxes in Figure 1) each bind to sites that do not vary among most bacteria, i.e. they are “universal” 16S DNA sequence domains. The resulting “Secondary Amplicon” is thus well suited as a target analyte for hybridisation to an array of DNA hybridisation probes, comprising a microarray derived from the well-known sequence diversity within the 16S region within the domain defined by labelling PCR.
The general location of such known 16S sequence diversity is identified in Figure 1 as “Microarray Probe Hybridisation Sites” (middle of Figure 1, in black) with the Sequence ID for several of those hybridisation probes, as originally specified in Patents 10,501,814 and 10,513,745 (in black) positioned close by to each location.
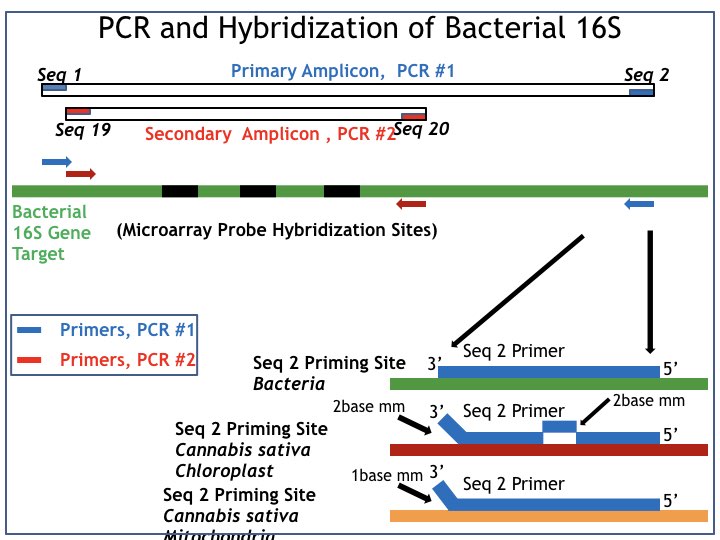
Figure 1: PCR and hyridisation of bacterial 16S
The same “universal” 16S sequence structure to which Seq 19 and Seq 20 each bind (red boxes) is also seen in the eukaryotic 16S-like sequence elements, which persist in the mitochondrial and chloroplast genomes of cannabis and hemp, and even in other plants1,2,3.
Consequently, in the presence of cannabis DNA contamination in a bacterial DNA sample, the eukaryotic 16S-like sequences will be amplified by a simple “universal” PCR reaction such as that driven by Seq 19 and 20. Such ambiguity is well-discussed in the referenced literature.
Cannabis microbiology
To eliminate the risk of specious cannabis “16S-like” DNA contamination in the PCR reactions, we have chosen to develop the assay as a two-step tandem PCR reaction series. In that series, a first PCR reaction is performed on the raw bacterial lysate (“loci enhancement” in Figure 1) to generate a “Primary Amplicon”.
The loci enhancement reaction is driven by the PCR primer pair Seq 1, Seq 2 (blue boxes), which are those described in Patents 10,501,814 and 10,513,745. Detailed sequence analysis reveals that Seq 1 (blue box, left) is common to both bacterial 16S and the cannabis eukaryotic “16S-like” sequences. Thus, it is Seq 2 (blue box, right) which confers the needed PCR sequence specificity.
The sequence structure of the reverse PCR primer for loci enhancement (Seq 2, blue box) is exactly complementary to the 16S DNA sequence of most bacterial species of interest in cannabis microbiology
The origin of the needed sequence specificity is elucidated in the lower right-hand corner of Figure 1. There, it is shown that the sequence structure of the reverse PCR primer for loci enhancement (Seq 2, blue box) is exactly complementary to the 16S DNA sequence of most bacterial species of interest in cannabis microbiology, i.e. “Seq 2 Priming Site, Bacteria” (top).
Conversely, the analogous “Seq 2 Priming site in Cannabis sativa Chloroplast” (middle) and the “Seq 2 Priming site in Cannabis sativa Mitochondria” (lower) have both been designed to display a well-defined mismatch at the 3’ terminus of the primer-template complex. Such 3’ terminal mismatches are well-known in PCR practice to render such primer-template complexes unable to support PCR under ordinary conditions of high specificity PCR thermal cycling.
The Seq 2 Priming site in Cannabis sativa Chloroplast (middle) is seen to additionally possess an internal two-base pair mismatch, which would provide for additional inactivation of its function as a reverse PCR primer for loci enhancement.
The combined use of Tandem PCR+Microarray hybridisation enhances the specificity obtained from the Tandem PCR pair alone. This change is because throughout evolution, the eukaryotic 16S-like sequences, in both chloroplast and mitochondria, have diverged significantly from those seen in modern bacterial species, especially within the region of high 16S sequence diversity, i.e. the “Microarray Probe Hybridisation Sites” identified in the middle of Figure 1.
Microbial discrimination
As a result of that substantial 16S sequence divergence over time, eukaryotic 16S-like DNA sequence obtained from amplification, of either C.sativa chloroplast or C.sativa mitochondrial DNA, don’t match, in most cases, with the sequence designs in place for the microarray probes specific for bonafide bacterial 16S DNA. Hence it provides for a second-tier discrimination beyond that obtained from just the Tandem PCR reactions (#1, #2), as demonstrated in Figure 2. We use Genomic DNA from Oklahoma Hemp Female, C.sativa strain 17965 and strain FL-1459), and E.coli, as well as NTC and PCR amplified using plant chloroplast primer set and bacteria (loci enhancement) separately.

Figure 2: Primer sets test on Cannabis and E.coli samples
Plant gDNA was amplified by the chloroplast primer set, while E.coli DNA was amplified by the bacteria primer set (labelled as “+”). The reciprocal amplification didn’t happen with the reciprocal primer set (labelled as “-”).
Data from multiple customer labs using well-characterised cannabis samples have confirmed the combination of bacterial 16S specific Tandem PCR, coupled to bacterial 16S specific microarray hybridisation, provides in the aggregate for highly specific bacterial detection and speciation. The latter is based on 16S DNA analysis under conditions where false-positive signals do not occur due to contamination with cannabis DNA. In Figure 2, we demonstrate that loci enhancement (Seq 1 and Seq 2) do not amplify residual “bacterial 16S-like” DNA in representative cannabis samples.
References
- Dorn-In S, Bassitta R, Schwaiger K, Bauer J, Hölzel CS (2015) Specific amplification of bacterial DNA by optimized so-called universal bacterial primers in samples rich of plant DNA.
- J Microbiol Methods. Jun;113:50-6. doi: 10.1016/j.mimet.2015.04.001. Epub 2015 Apr 9.
- Tsoktouridis G, Tsiamis G, Koutinas N, Mantell S (2014). Molecular detection of bacteria in plant tissues, using universal 16S ribosomal DNA degenerated primers. Biotechnol Biotechnol Equip. 2014 Jul 4;28(4):583-591. Epub 2014 Oct 22.
N.B. This article is featured in the February 2020 issue of Cleanroom Technology. Subscribe today and get your print copy!
The latest digital edition is available online.