There are currently differences between European and US testing methodologies for validating a disinfectant. The manufacturer must discern the regulators they need to comply with, and then the expectations of those regulators. To read more about the regulators and their standards, the article "The rules for validating a disinfectant".
The key part of validation is the efficacy testing and how it is carried out and that is what the focus of this article will be.
European efficacy tests
A facility that needs to validate a disinfectant in Europe has a structured series of tests devised by the CEN Technical Committee (TC/216) to use. This structure is highly accepted by member state GMP regulators.
The CEN disinfectant test methods are summarised in EN 14885:2018 9, a very helpful "Standard of standards" which specifies the European Standards to which products have to conform to claim microbial activity. It covers methods for vegetative bacteria, fungal spores, yeasts, virus and mycobacteria and bacterial spores and is applicable to products to be used in the area of human medicine, veterinary areas, and in food, industrial, domestic and institutional areas. It is currently under revision as pr EN 14885:2020 (E).
There are maybe only two disinfectants approved for use in both North America and Europe
Phase 1 tests: Quantitative suspension tests to establish that a product under development has bacterial, fungicidal, yeasticidal or sporicidal activity without regard to specific areas of application.
Phase 2 comprises two steps: Step 1 tests are quantitative suspension tests to establish that a product has bacterial, fungicidal, yeasticidal, mycobactericidal, tuberculocidal, sporicidal, virucidal, activity under simulated practical conditions appropriate to its intended use. These tests prove the irreversible inactivation of microorganisms. But the activity of the product is against microorganisms in suspension and desiccated microorganisms might be stressed and pose different challenges. Step 2 tests are quantitative tests to establish that a product has bactericidal, fungicidal, yeasticidal, mycobactericidal, tuberculocidal, sporicidal, virucidal, activity when applied to a surface under simulated practical conditions. Phase 2, step 2 tests provide information against desiccated microorganisms on inanimate surfaces
Phase 3 tests are field tests under practical conditions. The applicable methodologies for this type of test are not yet available but may be developed in the future. Guidance on the design of phase 3 tests and the use of data from phase 3 tests is now provided in Annex C of EN 14885.
Tests must be carried out under the minimum / obligatory conditions if specified in the standard. Additional test organisms, temperature, contact times and interfering substances can also be carried out but must be specified. Generally, both Phase 2, step 1 and Phase 2, step 2 tests are needed in combination to support efficacy claims.
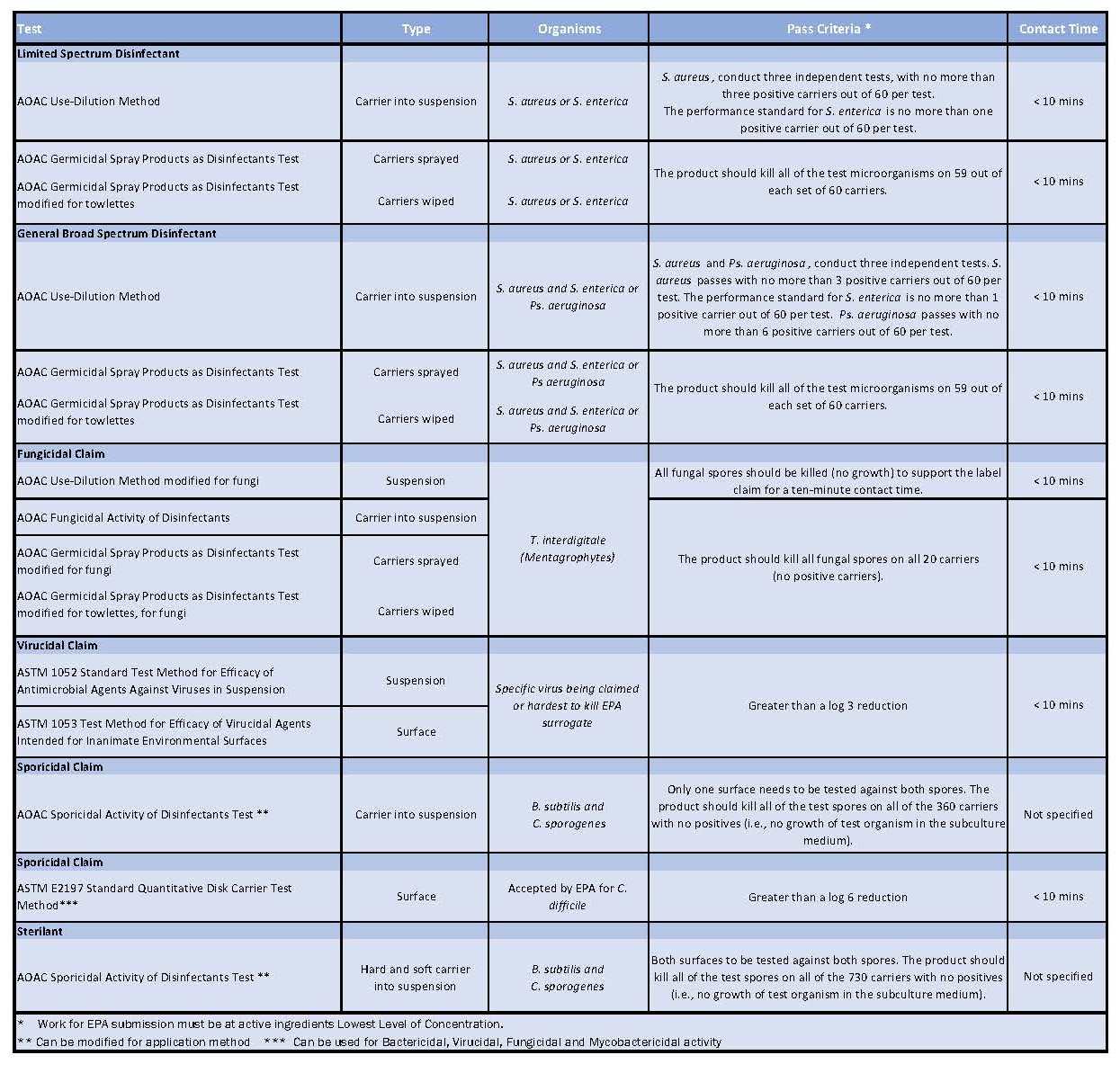
Table 1: Showing all the test methods useful for testing disinfectants in cleanrooms
There isn't currently a Phase 2, step 2 test for testing disinfectants with mechanical action in industrial areas (one is currently in progress) but there is one available for the medical area so this is used. Medical area tests are also used for virucidal testing. There is a provision to use a standard from another area if one doesn't exist, modifications can also be made but the modification has to be declared and state the product was tested in accordance with the principles of the "X" test.
The standards are routinely updated or amended and this can be seen by the date following the standard number, Eg EN 13697:2015+A1;2019.11.
The foreword of the standard will state if any changes have been made which might impact the results from a previous version. "The changes mentioned above have no impact on the test results obtained with reference to the previous version. Those results are still valid." Or "The changes mentioned above have an impact on the test results obtained with reference to the previous version. Products have to conform to this new version in order to support the claims for the activity corresponding to this European Standard". Be aware when looking through manufacturers data as very often results will just be declared to a particular test without the standard's date being specified. This was especially relevant when "spiny spores" were added to EN1650 12 and EN13697 11 test methods. Disinfectants which had passed the previous tests, would not always pass the new test method. 70% IPA solutions are an example of this.
United States efficacy tests
While any antimicrobial product could be validated, most facilities use products that are already EPA registered as disinfectants. The pass/fail criteria for cGMP disinfectant validations are considerably different than for EPA registration as a disinfectant.
A facility may choose to validate with more dilute versions of concentrates or faster contact times. The procedures used to validate should be scientifically sound and often are based on existing standards, but with opportunities to modify (e.g., lower inoculum, no soiling).
EPA registration requirements do not address how disinfectants are used in pharmaceutical, biotechnological, and medical device industries. Manufacturers within these industries are instructed to validate disinfectants by demonstrating efficacy through the following analyses:
Use-Dilution tests against a wide range of standard organisms and environmental isolates specific to a particular location.
Pass/fail criteria for cGMP validations are considerably different than for EPA registration
Surface challenge tests using standard organisms and typical environmental isolates specific to a particular location, applying disinfectants to surfaces at the specified concentration and contact time.
Statistical comparisons of the frequency of isolation and number of microorganisms isolated before and after implementing a new disinfectant or protocol.
It is worth bearing in mind especially when reviewing the literature from a US disinfectant manufacturer that the definitions for sanitiser, disinfectant and sporicide differ in the US from those commonly used in Europe.
The EPA defines a sterilant as something used to eliminate or destroy: fungi, fungal spores, viruses, vegetative bacteria, bacterial spores and can be a liquid chemical sterilant used by immersion. A sterilant is used to inactive spores on both hard and soft inanimate surfaces. The product should kill all of the test spores on all of the 720 carriers with no positives against (AOAC) Official Method 966.04 Sporicidal Activity of Disinfectants test. A sporicide is used to inactivate bacterial spores on a single chosen inanimate surface, the product should kill all of the test spores on all of the 360 carriers with no positives against (AOAC) Official Method 966.04 Sporicidal Activity of Disinfectants test.
Table 1: Showing all the test methods useful for testing disinfectants in cleanrooms
Disinfectants are defined as something used on nonliving surfaces and objects to destroy or irreversibly inactivate infectious fungi and bacteria but not necessarily their spores. A broad-spectrum disinfectant has a label claim against both Gram-negative and Gram-positive bacteria, a limited spectrum disinfectant has activity against one but not both. A sanitiser is defined as a product that is used to reduce but not necessarily eliminate microorganisms from the inanimate environment to levels considered safe by public health codes or regulations.
The validation of disinfectants in cGMP environments in the United State is conducted with a variety of official and non-official methods using guidelines from for example USP <1072> 13, PDA TR#70 14 and other documents. USP <1072> goes on to state that disinfectant efficacy studies must have realistic acceptance criteria, at least 2-log reductions for bacterial spores and 3-log reductions for vegetative bacteria.
Validation procedures are often based on existing standards, but modified
It is very clear from the GMP regulations that pharmaceutical manufacturers need to design, validate and implement, a documented and approved disinfectant programme as part of their contamination control strategy.
There are many standard disinfectant efficacy test methods available in both Europe and the United States but the testing approach is very different and using the tests in the opposing regions is not so easily accepted. Currently, there are very few, maybe only two disinfectants, which are approved for use in both North America and Europe, available with a data package of both EN and AOAC / ASTM testing. Guidance documents are available to help create the disinfectant validation plans for an aseptic facility and disinfectant manufacturers and test labs can help with the design and implementation of a disinfectant efficacy testing programme.
