
Understanding different types of contamination, where contamination might originate from, and how contamination can be transferred, represents an important area of quality assurance in relation to the manufacture of pharmaceutical and healthcare products. Approaching this assessment in a stepwise and logical manner, drawing on scientific and technical principles, increases the success of such assessments. The need for such assessments and their importation into an overarching ‘contamination control strategy (CCS)’ is a cornerstone of the EU GMP Annex 1, Manufacture of Sterile Medicinal Products, Version 12 draft revision process.
The strategy includes an assessment of areas such as:
- Microbial contamination
- Viral contamination
- Chemical contamination
- Sterility assurance
- Cleanroom design
- Facility layout and process flows
- Particle contamination (both visible and subvisible)
- Cross-contamination
- Design of primary and secondary packaging
There are different areas that require assessment, and each assessment completed helps to layer and strengthen the CCS. One fundamental area is with the cleaning and disinfection of cleanrooms.
The update of Annex 1 of EudraLex “The Rules Governing Medicinal Products in the European Union” helps to structure some of the aspects that need to be examined, not least by drawing an acute distinction between ‘cleaning’ and ‘disinfection’.
Yet even with the expanded guidance, approaching a review of cleaning and disinfection is not straightforward. There are multiple areas to consider and these need to be ordered and worked through sequentially for an assessment to be effective and thus for contamination risks to be addressed.
One way to assess these risks is by using a new digital tool from Ecolab Life Sciences. The approach is based on a combination of audit, data inputs, intuitive software, and an unambiguous output. With the case study in this article, there were two areas of concern: transfer of items and the presence of disinfectant residues.
The Ecolab approach
Ecolab has developed a digital Contamination Control Strategy assessment tool. Through the proprietary software, an Ecolab auditor in conjunction with the facility are able to review the current state of cleanroom cleaning and disinfection against regulatory requirements and best practices. The tool takes the reviewers through a stepwise assessment of cleanroom operations, comparing these against the requirements of global regulations and best practices.
This is undertaken by breaking down sections of operations and providing a risk framework (such as the process for introducing non-routine items into the cleanroom). The information is captured digitally and output provides both an overall risk score and, where gaps are identified, the opportunity to mitigate risks.
Case study: Improving transfer disinfection and residue management
To test out the Ecolab approach, a study was undertaken at Bio Products Laboratory in the UK. This is a large manufacturing facility, with processing occurring across over 200 cleanrooms of EU GMP Grades D to B, and with sterile filling occurring within Grade A zones. While the facility makes use of autoclaving and has the capability to automatically decontaminate many items by gaseous hydrogen peroxide (35% w/v), many items still need to be prepared through a transfer hatch.
The objective of undergoing the Ecolab assessment and subsequent analysis, was to look for any additional areas that could lead to contamination events as part of an on-going commitment to continual improvement. While most areas assessed were deemed satisfactory, there were two areas for improvement.
Transfer disinfection
No matter how well designed a cleanroom is, it remains at risk from microbial contamination in the transfer process and such risks are arguably exacerbated when people are involved. A proactive response is based around cleaning and disinfection. Operating an effective cleaning and disinfection regime requires thought and planning, and some situations present greater challenges.
It has long been recognised, as indicated in several regulatory findings, and given due consideration in EU GMP Annex 1 Version 12 draft, that the transfer of items into the cleanroom represents a contamination vector. This is not least because the outer packaging of all items will contain microorganisms.
The contamination severity becomes greater as an item moves through the cleanroom classification cascade; this is because the number of permitted microorganisms diminishes meaning that the risk presented by survivors becomes greater. In response to this severity, measures need to be taken to reduce the likelihood of contamination transfer. Such measures include the use of autoclaves or, for single-use sterile items, automated decontamination chambers. However, many items are not suitable for autoclaving (together with the industry trajectory towards the use of single-use items) and many facilities do not have the capacity for automated processes. This means there is a continuing reliance upon the transfer hatch and the transfer disinfection process.
Even with recently expanded guidance, approaching a review of cleaning and disinfection is not straightforward
With this reliance comes the risk of microbial contamination transfer, especially with spore-forming microorganisms due to their greater resistance to cleaning and disinfection processes and their ability to survive for prolonged periods (especially in the case of bacterial endospores) under non-ideal and harsh environmental conditions.
With the transfer of items into a cleanroom, the greatest challenge and area to avoid is with the transfer of bacterial spores into the Grade A environment. This presents a significant risk of contamination to aseptically prepared products and potential patient harm. An important step for reducing this likelihood is with preventing spores being transferred from Grade C to Grade B as items are moved through a transfer hatch. When disinfecting, it is important to:
- Physically remove the bioburden from the surface
- Ensure the presence of sufficient disinfectant for long enough to kill microorganisms
- Facilitate the destruction and removal of contaminants by the mechanical action of wiping, which enhances the biocidal effect through entrapment and physical removal
In the assessment, the process flow is reviewed and this needs to begin from the point of delivery to a warehouse and assess the types of packaging and wrapping, where this is removed, and the stages where disinfection takes place. This risk-based approach also considered the type of disinfectant required. Figure 1 provides a simplified summary of the process in use at BPL.
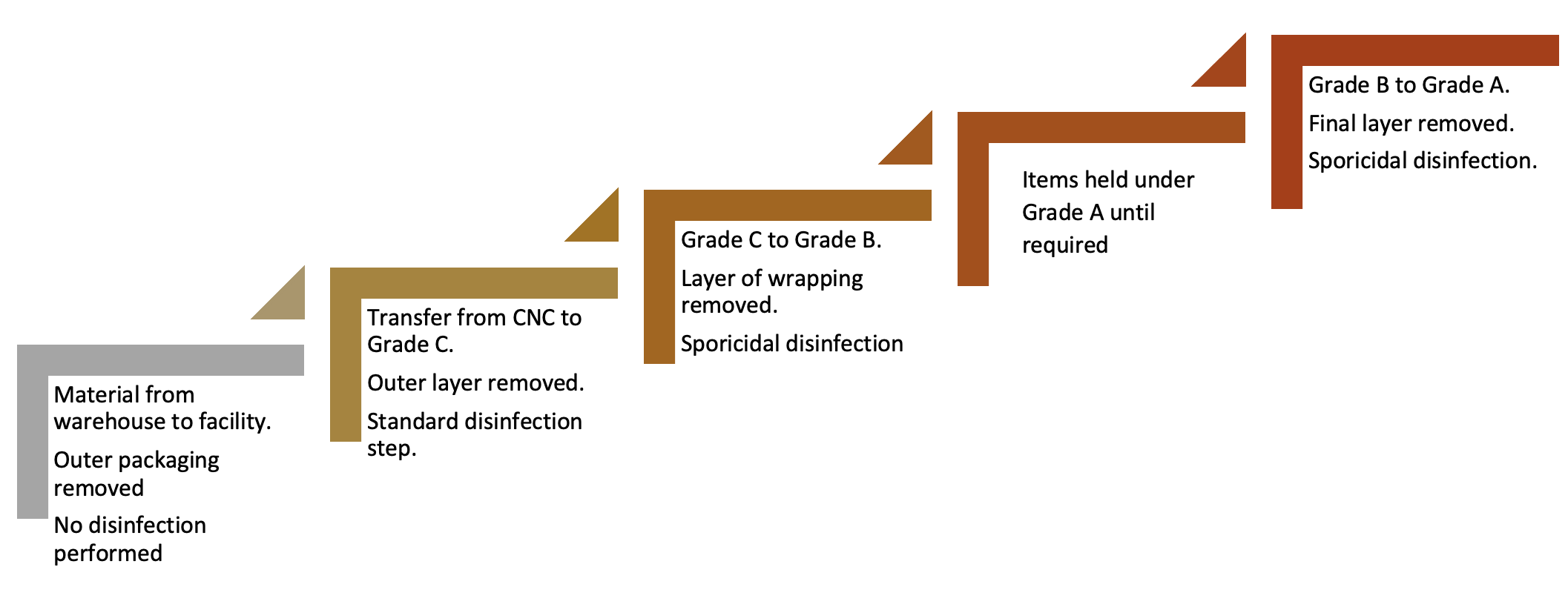
Figure 1: Simple process flow of material transfer
Items held under Grade A until required
With the transfer disinfection concept, a series of steps are required. This is because no single step completely addresses the bioburden risk and also because bioburden can arise at different points along the process. Contamination can arise from the external environment, the internal environment, surface cross-contamination (such as from cart surfaces), and via personnel. Ensuring appropriate levels of education and training of staff can help to reduce transfer risks, especially from gloved hands.
While the process at the facility was robust, there were some areas of weakness:
A. Due to risk that outer packaging presents from delivery from the warehouse to the facility, including the requirement for personnel to remove this, a sporicidal application was deemed to be good practice for addressing the risk of spore contaminants which are typically prevalent in these areas.
B. While the transfer from Grade C to Grade B was theoretically effective, and involved the use of a sporidical agent, surface environmental monitoring had not been undertaken to assess the pre- and post-disinfection bioburden challenge.
Both of these recommendations were relatively straightforward to accommodate and led to a strengthening of contamination control.
Residue management
Many disinfectants leave residues on surfaces. Residue profile is a combination of the amount of active ingredient and, where relevant, of inactive ingredients added to stabilise or boost the active compound. The types of residues that are most common are non-volatile residues (such as dissolved inorganic materials, e.g., sodium, anions, cations, or organic material).
Disinfectants that contain additional surfactants can also be more prone to forming residues. The presence of residues can also relate to the application technique. The choice of wipe/mop and the smoothness of the surface play an important role. Uneven application can lead to a concentration of liquids. Similar problems can arise from oversaturation of the surface. The unmanaged presence of residues within the cleanroom creates several problems (5-7):
- Product contamination can occur. This can be within vessels or where product contact items are disinfected, particularly in early stage manufacturing when items are not required to be sterile. Furthermore, surfaces recently disinfected present a risk where residues can be carried by operators (on their gloves or gowns) from one activity to another.
- Residues can render surfaces slippery, leading to health and safety risks.
- Residues can lead to surface damage over the longer-term, such as causing erosion, pitting and rouging. This will particularly occur in aged surfaces where surface depressions exist. Surface damage as a result of residue formation can contribute to biofilm development.
- Residues can leave surfaces sticky. This can aid the physicochemical interactions that bind microorganisms to surfaces. An essential feature of cleaning and disinfection is with the disassociation of as many microorganisms from surfaces as possible, since organisms in the planktonic state are easier to kill than those in the sessile state.
- Residues may contribute to the shielding of microorganisms from future applications of disinfectants.
- Residues that build-up and which are then resuspended can lead to particle generation.
- Residues of one disinfectant can interact with another. This mixing of chemistries can lead to a loss of efficacy of the active ingredient and therefore the effectiveness of the disinfectant is decreased and no longer representative of the initial validation.
- The presence of residues can be challenging for environmental monitoring. While contact.plates should contain neutralisers these are generally effective against low levels of residues. A high residue gradient may overcome the neutraliser and lead to an environmental monitoring false negative.
Assessing and addressing residues is required as part of EU GMP Annex 1 compliance (EU GMP Annex 1, Manufacture of Sterile Medicinal Products, Version 12 draft states that: “Cleaning programs should effectively remove disinfectant residues”) (8). The primary way to assess residues on surfaces is through visual examination. Since the same residue can appear differently on different surface materials, it is important that cleaning staff are appropriately trained.
With the case study, there had not been a detailed assessment of the risk of residues within the facility and therefore the Ecolab assessment provided the means to assess these risks and address them. Two ways were proposed to address disinfectant residues. One is to rinse areas following the application of a disinfectant, either using alcohol (such as 70% IPA) or water of sufficient purity (such as WFI). As a minimum, the frequency of residue removal should be between disinfectant changes as part of the rotational pattern. Increased frequency may be required dependant on the residue profile of the disinfectants in use.
Implementing low residue disinfectants, where the manufacturer can provide scientific evidence that any residue is not long-lasting or insufficient to cause any of the points listed above significantly reduces the residue risk and brings operational efficiency by reducing rinsing steps. The thorough assessment of residues has led to a revised strategy for the controlled management of disinfectant residue contamination risks.






