Life science cleanroom facilities must know that the disinfectants they are using can achieve effective levels of microbial kill across a range of surface types. There are many different types and formats of disinfectants now available for use within pharmaceutical cleanrooms and there are various national and international efficacy testing standards available to compare and qualify them.
The various standards and methods available to qualify disinfectant efficacy use different methodologies and have also not been written specifically for cleanrooms so the microbial test expectations are not always suitable. There are currently differences between EU and US testing methodologies. The expectation of different regulators such as the US Environmental Protection Agency (US EPA) and the European Chemicals Agency (ECHA), who are responsible for the safety of disinfectants/biocides placed on their respective markets, and the regulators responsible for the safe manufacturing of medical products such as the FDA and the MHRA, all differ.
Who’s in charge
Disinfectant validation for pharmaceutical cleanrooms includes many factors, including but not limited to, wet contact time (in vitro and in use) unopened and in-use shelf life, sterility, residues, application methods, storage, disposal, health and safety, audit of manufacturer, as well as the key factor of proving that the disinfectants being used can achieve the required levels of microbial kill on the key surfaces.
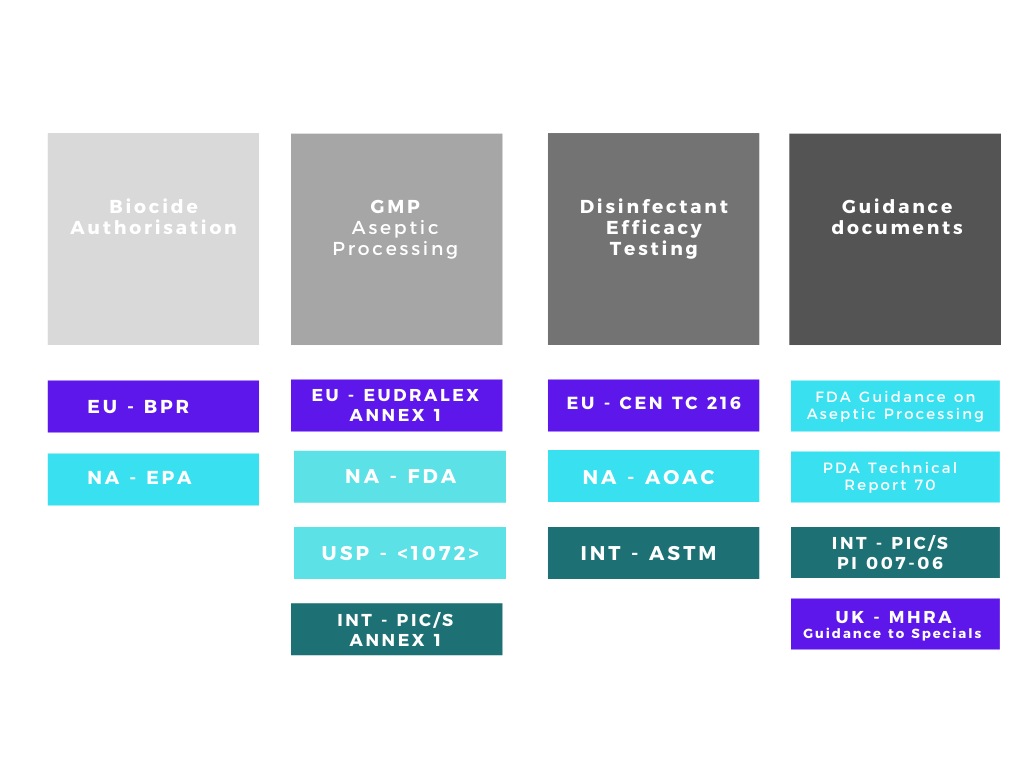
There are many acronyms that will be used when discussing disinfectants and their validation, FDA, EPA, BPR, AOAC, EN, EU GMP, USP, ASTM the list goes on! The regulatory web can get quite confusing especially if disinfectants are being qualified for use in different countries. We can simplify the web by separating out the regulatory and testing bodies into four main groups:
1. Regulatory bodies responsible for making sure biocides / disinfectants placed on their respective markets are both safe and efficacious.
2. Regulatory bodies responsible for ensuring the safety of medicinal products in their respective markets.
3. Organisations with approved testing methods for disinfectation efficacy qualification
4. Supporting organisations with guidance documents
Disinfectant/biocide authorization
In many, if not most countries, disinfectants must be authorised by the relevant regulatory authority before a manufacturer/supplier can place the product on the market. In the EU and the UK, all biocides must be authorised for sale under the EU Biocidal Products Regulation (BPR) and the GB Biocidal Products Regulation (GB BPR) respectively.
In the US, disinfectants, sanitisers, and sterilants are regulated by the Environmental Protection Agency (EPA) under the authority of the Federal Insecticide, Fungicide, and Rodenticide (FIFRA) Act. 2.
Neither EU GMP nor US FDA refer specifically to disinfectant efficacy test standards
European Union
Since September 2013, national biocide registrations in the EU have been replaced by one common approvals process (EU No 528/2012) the Biocidal Products Regulation. The BPR is implemented by the European Chemicals Agency (ECHA). All biocidal products require an authorisation before they can be placed on the market, and the active substances contained in that biocidal product must have been previously approved. The BPR is currently an on-going process so national registrations apply until the active substance used in a biocidal product is approved at which point a disinfectant manufacturer has 18 months to submit a dossier for authorisation in all member states they wish to sell.
It is very clear within the regulation that it is the “intent of a formulation which determines its definition as a biocide”. The absence of a claim on either literature or packaging does not mean the product is not a biocide and doesn’t need to be authorised. It costs many 1000’s of euros to provide all the information required for biocidal authorisation and submit for Union (all countries) authorisation or authorisation in multiple countries. Not all cleanroom disinfectants forms, which includes presaturated wipes, have been submitted for approval due to the high costs involved. The format of the disinfectants doesn’t matter, whether it is in a trigger spray, presaturated or saturate at point of use wipe, a two part system which needs to be mixed, or that it is being gassed or fogged it still needs to be authorised in the manner in which it will be used.
Some examples of active ingredients commonly seen in cleanroom disinfectants are IPA and denatured ethanol hypochlorites, hydrogen peroxide, peracetic acid, quaternary ammonium compounds and phenols.
The ECHA published a guidance document for suppliers “Guidance on the Biocidal Products Regulation Vol II, Efficacy 3”, which gives detailed information on what test work needs to be submitted to show a products efficacy. This also forms a really good basis for any cleanroom facility starting disinfectant efficacy testing.
It states that standard test methods and standard species should be used. In addition, only quantitative test methods should be used, and it recommends that a tiered approach to testing is followed. Additional test organisms can be used but only if their relevance is justified and the relevant test must also be passed with standard organisms. Both the site and method of application must be specified, ie wiping in a cleanroom. The testing of the product should be appropriate for the application method, a suspension test could only be used if the surface to be disinfected was to be immersed for instance. Test methods do exist for surface testing both with and without mechanical action.
The regulatory web can get quite confusing
ECHA recommends that a manufacturer ideally uses internationally recognised test methods such as CEN, OECD or ISO. The use of CEN test methods is highly recommended, if no CEN test exists then OECD test methods maybe used. Other test methods are available eg AOAC, ASTM, US-EPA, VAH, AFNOR but these can only be used when there is no international standard available for a specific application.
With the introduction of disinfectant efficacy test methods from CEN Technical Committee (TC/216) a wide range of harmonised European methods for testing the activity of disinfectants used in medical, veterinary, food, industrial, domestic and institutional areas are available. TC/216 proposed a three phase evaluation process for disinfectant efficacy testing. The ECHA states that existing guidelines can be modified to make them more suitable for the specific product or conditions, for example contact time, soiling but it must be clearly described and justified. Similarly, test conditions can be modified if the claimed use of the product is different from the obligatory test conditions.
When carrying out the testing, consideration should be given to temperature, contact time (must be practical in real life), neutralisation validation, pH, texture of surfaces, repetition of tests, pass criteria and the need for an interfering substance. In this instance, ECHA recognises that cleanrooms are a special case and that even the clean conditions of the EN test methods are overkill and WFI can be used as the interfering substance. The guidance also makes specific comment about testing disinfectant wipes, Phase 1, step 1 test should be carried out with fluid extracted from the wipe and phase 2, step 2 tests should be done with mechanical action.
United States
Under the Federal Insecticide, Fungicide, and Rodenticide (FIFRA) Act, any substance that prevents, destroys, repels, or mitigates pests, including microorganisms, is considered a pesticide. Chemical disinfectants are considered antimicrobial pesticides. A disinfectant manufacturer in the US must register their product including fees with the EPA, and with every individual state they wish to sell in. A product is approved when the EPA concludes that it is safe and effective when used as directed by the label, which includes chemical characterisation, safety, and efficacy data.
Data demonstrating the efficacy claim of a disinfectant, whether it is bactericidal, fungicidal, sporicidal or viricidal, is a clear requirement of the EPA for a disinfectant manufacturer to achieve registration. Some key points expected by the EPA are that the data should be generated in compliance with Good Laboratory Practice Regulations (GLP) and that disinfectant manufacturers, test products for biocidal efficacy at the active ingredients’ lowest level of concentration (LCL). Guidance for manufacturers is provided by the EPA’s Product Performance Test Guidelines; OCSPP 810… details the test methods to be used by disinfectant manufacturers to support claims of microbiocidal activity.
Disinfectant validation for pharmaceutical cleanrooms includes many factors
In a similar manner to the BPR, the EPA recommends specific tests using specific reference microorganisms and product application techniques to substantiate disinfection claims. The Product Performance Test Guidelines include test methods from both the Association of Official Analytical Chemists (AOAC) and American Society for Testing and Materials (ASTM). The EPA has long preferred so-called "hard surface carrier" methods for substantiation of efficacy claims. Test surfaces called “carriers” are inoculated, dried, and then treated with the disinfectant. Most hard surface carrier methods are qualitative, meaning pass or fail determinations are made based on whether one or more test microorganisms survive treatment with a disinfectant. Some other methods are quantitative, meaning percent reductions are calculated by counting viable microbial populations before and after treatment with the disinfectant.
Good Manufacturing Practice
Pharmaceutical companies selling medicinal products in the EU are required to implement the necessary measures in order to comply with the requirements set out in EudraLex Volume 14 of the “Rules Governing Medicinal Products in the European Union - EU Guide to Good Manufacturing Practice. Pharmaceutical companies selling medicinal products in the United States are required to comply with the GMP requirements of Title 21 FDA CFR 211. Current Good Manufacturing Practice for Finished Pharmaceuticals.
Part of these necessary measures include the design, validation and implementation of a documented and approved disinfectant programme. It will form a key part of any pharmaceutical production area qualification.
The current draft of Annex 1 of EU GMP, 7 specifically covering sterile medicinal products has an expectation that cleaning and disinfection forms part of a facilities contamination control strategy. Its states that the disinfection process should be validated. Validation studies should demonstrate the suitability and effectiveness of the disinfectants in the specific manner in which they are used and should support the in-use expiry of prepared solutions. The US FDA Guidance for Industry, Sterile Drug Products Produced by Aseptic Processing (Aseptic Processing Guide September 2004) 8 states that each manufacturer must have a formal programme governing the qualification, use and disposal of disinfectants. The suitability, efficacy, and limitations of disinfecting agents and procedures should be assessed. The effectiveness of these disinfectants and procedures should be measured by their ability to ensure that potential contaminants are adequately removed from surfaces.
However, neither EU GMP nor US FDA guidance make any specific reference to disinfectant efficacy test standards. Neither mention the pass criteria that need to be achieved for an individual disinfectant, the decision on level of microbial kill will be based on room grade, environmental monitoring, risk assessment and form part of the contamination control strategy for the facility. But both mention the importance of the pharmaceutical manufacturer evaluating the efficacy of disinfectants used both, in the manner in which they are to be used and on the surfaces they will be used. Regulators in both Europe and NA will expect to see disinfectant qualification and validation.
Even though a cleanroom disinfectant needs to be authorised/approved with the relevant biocidal registration system, the GMP regulations do not specify that this has to be the case. However, using an authorised disinfectant gives reassurance of continuity of supply (an unauthorised disinfectant could be pulled from the market) and independent health and safety and efficacy information.
