Contamination prevention is a critical fact in pharmaceutical production. The new EU-GMP Annex 1 (Guideline for manufacture of sterile medicinal products) therefore contains the reference to a “Contamination Control Strategy” (CCS) without, however, defining its exact content. Without precise and detailed process knowledge, which is a decisive factor, the wrong conclusions are often drawn. One example of this is the overestimated criticality of room pressurisation.
When a pharmaceutical company opens a new factory, the "state-of-the-art" status is always highlighted. In most cases, this status is more than 50 years old and does not stand up to any scientific justification, and should be seen as a rule of thumb. These include, for example, extremely high air exchange rates, closed processes in an unnecessarily high cleanroom grade, and room pressure that is considered indispensable.

Figure 1
Room pressurisation steps at a level of 0.05 inch of water column (~ 12.5 Pascal Pa) was arbitrarily defined in the 1950s and is still the reference value up to date with the mistaken opinion the higher the value the better the prevention of infiltration of contaminants. At a level of 0, 5, 10, or up to 20 Pa technical investigations do not show significantly higher prevention.
The value of 10 to 15 Pa is readily achievable, easy to monitor, and appears to prevent contamination transfer from adjacent rooms or the environment without knowing the real way of contaminants. One main fact is always ignored: it is a physical law that at a differential pressure of 0 Pa there is no flow and differential pressure at a value of >0 Pa between cleanrooms is only valid until the door is opened.
A room overpressure at the level of 10 to 20 Pa can never create sufficient airflow between two rooms with doors open. At that point, through the door-opening process or people and carts moving between rooms, there is always an exchange of air that facilitates contamination although a room overpressure is seeming to be present.
Room overpressure is the most overrated cross-contamination prevention tool, and this article should help better understand the actual effect and impact on a production line inside a cleanroom.
Understanding the way contamination occurs
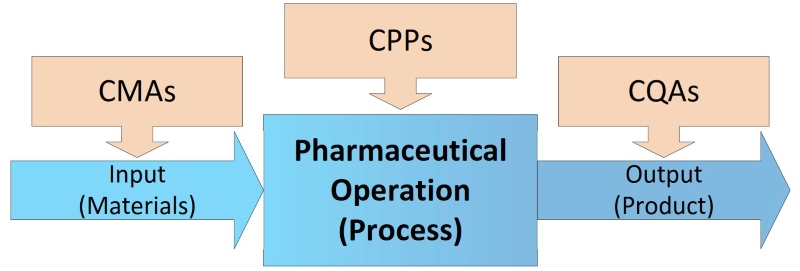
Figure 2
To prevent contamination and cross-contamination, process developers must identify sources of potential risk in all production steps with Critical Material Attributes (CMA), the influence on Critical Process Parameters (CPP), and Critical Quality Attributes (CQA) of the final product (see Figure 2). Within the CCS the current controls and actions that will be taken to eliminate and control them have to be defined. Sources of contaminants like particle matter as non-viable (NVP) and microorganisms (VP, viable – bacteria, yeast, mould), are very different and could be, but are not limited to:
- Personnel with gowning and behaviour
- Personnel and material flow
- Surfaces with a product contact
- Material and packaging components
- Outside air with particle and chemical contaminants
- Utilities like water and compressed gases
- Facility construction and finishes
Airborne Molecular Contamination (AMC) is not seen as a generally critical source in pharmaceutical production up to date. Contamination pathways could appear during direct exposition (open product) to the environment by contaminated air with high turbulence caused by an excessive air-change rate (ACR), surfaces with product contact, starting materials, and utilities like water or compressed gases.
Transfer contamination appears by mixing of products, carryover, transmission, indirectly via auxiliary devices, means of transportation, and last but not least by personnel. Room pressurisation could have an impact only on airborne suspended particles which infiltrates through leaks in the enclosure of the cleanroom and this appears typically not in the centre of the cleanroom near the processing line.
The control of the specified room pressure requires a certain leakage of cleanroom enclosure walls which contrasts with the specification of a tight cleanroom. The laws of physic say clearly that airflow through these leaks will only appear when there is a pressure difference, a different energy potential. This is why at a pressure difference of 0 Pa there will be no flow!
The impact of room pressurisation
Let’s take an example: a cleanroom of an EU-GMP grade C (ISO 8, max. allowed particles of 3,520,000/m³ at ≥ 0.5 µm) with a length of 16 m, a width of 6.25 m, and a height of 3 m have a cubic content of 300 m³.
The calculation of the needed air-volume flow results in 2,700 m³/h and is equal to an air-change rate per hour (ACR, ACPH) of 9-fold. I am sure that most of you have expected a higher ACR of about 20-fold or higher as the recommended (but always seen as required) “state of the art” value by the US Food and Drug Administration (FDA – Guidance for Industry, Sterile Drug Products Produced by Aseptic Processing, cGMP).
Recently, cleanrooms are increasingly defined by their tightness, and the german guideline VDI-2083-Part 19 (VDI Verein Deutscher Ingenieure, Association of German Engineers) contains recommendations for the tightness of cleanroom fabrics. Which individual rooms, room groups, with or without door gaps, and systems of a building must meet the airtightness requirements are to be determined by the planner or specified in the User Requirements Specification (URS). Keep in mind, the more airtight the room envelope, the more difficult it is to control a defined setpoint for the room differential pressure.
The room pressure control concept should be adapted to the tightness of the room, otherwise undesirable fluctuations in the room differential pressure can arise. For a grade C cleanroom, the air permeability should not exceed a value of 1.236 m³/h for each square meter of cleanroom enclosure at an overpressure of 50 Pa.
A room overpressure at the level of 10 to 20 Pa can never create sufficient airflow between two rooms with doors open
The above-mentioned example room has a surface area (including the ceiling, excluding the floor which should be tight) of 233.5 m² and allows therefore a permeability of max. 288.61 m³/h at 50 Pa. Using the “fan law” half the volume flow results in a quarter of the pressure loss – so at 12.5 Pa (a quarter of 50 Pa), the maximum leakage volume flow is thus calculated 144.3 m³/h (the half of 233.5 m³/h) and is about 5% of the ACR of 9-fold and becomes less when the pressure decreases and goes to zero at 0 Pa.
In the case of a negative pressure of -12.5 Pa, this amount of air (144.3 m³/h) would flow into the room from the adjacent rooms, in the worst case cleanrooms with a lower cleanliness class, and maybe with a higher particle load according to the cleanliness grade (e.g. grade “D” or lower). On the other hand, investigations on the subject of “human as particle source” show, that one single person in cleanroom garments (overall, hood, booties, gloves) without any activity, only standing, emits on average 28,800 particles ≥0.5 µm, 330 particles ≥5.0 µm, and approximately 18 germs every minute!
If we compare the particles and germs discharged by the personnel, the influence of the “contaminated air” flowing in through the leaks only in the case of negative pressure is a negligible value, while about up to 90% of microbial contamination could come from personnel. The good design of the air-locks and a well-considered clothing concept could be much more effective.
The direction and flow of air have a much more vital role in a CCS. Effective air ventilation with low turbulences in the cleanroom and flow from critical areas to areas with low contamination risk for the process/product is much more important and would easily and immediately flush away these “contaminants”. In most cases, processing will be done in the centre of the cleanroom and not near the walls and in the case of a closed process, it would certainly not affect product quality.
Conclusion
The room overpressure has a minor influence on cross-contamination and therefore does not justify the investment and operating costs in pharmaceutical cleanroom production. The investment includes the measuring devices (Figure 1) as well as the additional air-flow control valves with actuators, the data points for the building management system (BMS), and the additional costs for monitoring devices, commissioning, balancing, and qualification.
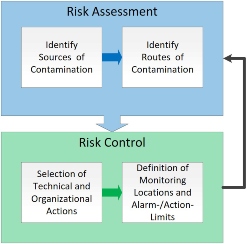
Operating costs include not only the requalification of the monitoring system with its devices and data points but also the internal costs of the resulting unnecessary editing of deviations on the internal not scientifically justified range of room pressure and pressure cascades. In the end, it pays to draw the right conclusions from the risk assessment (Figure 3) for contamination control.