This article provides an overview of the essential elements, including disinfectant validation and efficacy testing. To do this, we will look at regulatory requirements, industry standards, and best practices.
Regulatory background
For the US, cleaning and disinfection in pharmaceutical facilities are governed by US cGMP (21 CFR 211), EU GMP Annex 1 (2022), and industry guidance (USP <1072>, IEST RP 18.5), which emphasise validated methods for aseptic areas and real-world field trials.
The EU's Good Manufacturing Practices (EU GMP) Annex 1, revised in 2022, further elaborates on the requirements for disinfection and bio-decontamination. It highlights the importance of validated decontamination methods for barrier systems such as isolators and Restricted Access Barrier Systems (RABS), as well as the thorough cleaning and disinfection of cleanrooms.
Industry guidance documents, such as the U.S. Pharmacopeia (USP) <1072> and Institute of Environmental Sciences and Technology (IEST) RP 18.5, also offer additional guidance on designing and validating disinfectant efficacy studies. These documents recommend the use of in situ disinfectant field trials to ensure the effectiveness of cleaning and disinfection programmes in real-world conditions.
But what are the specific standards and guidelines that form the foundation of effective cleaning and disinfection validation programmes? Table 1 summarises the key areas within these standards.
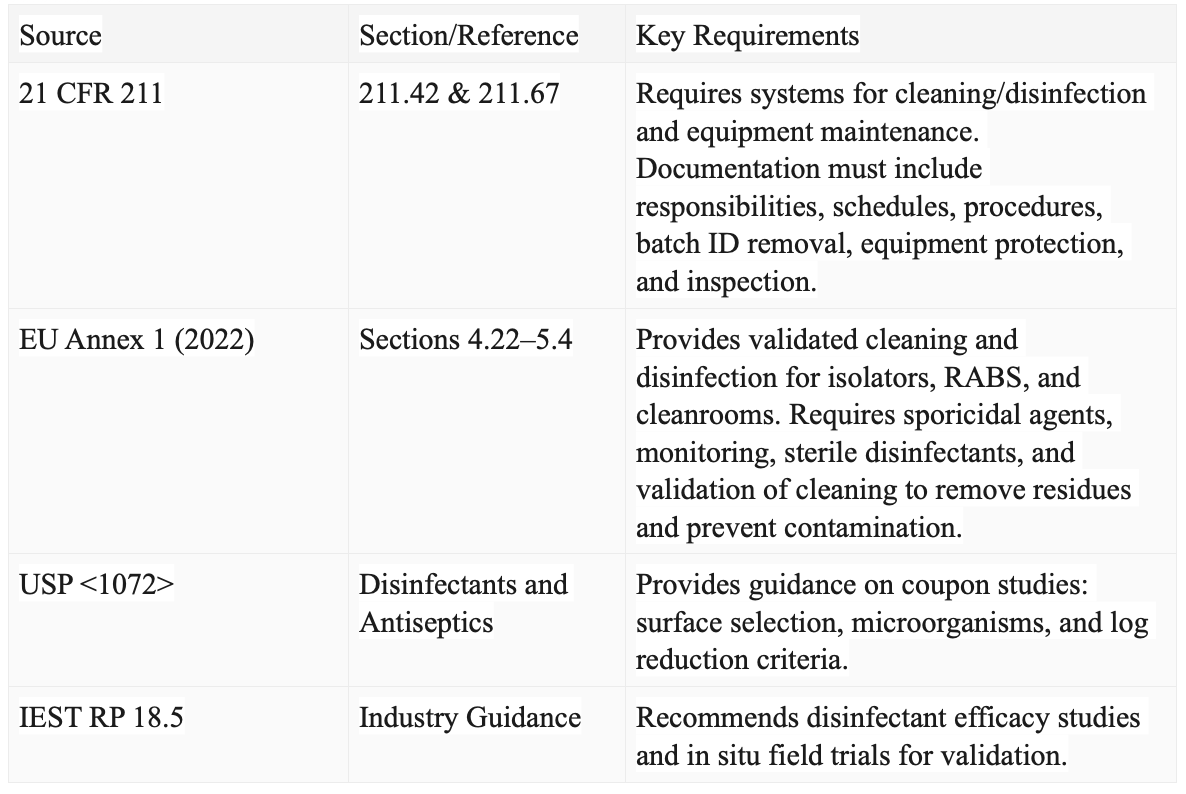
Table 1: Summary of Cleaning and Disinfection Validation Program Standards and Guidelines
Choice of cleaning, disinfectant, and sporicidal agents
Cleaning: For cleanrooms and critical environments, cleaning agents can be used for periodic residue removal or after a worst-case event (such as product spillage or construction) to bring the cleanroom back online. Many disinfectant formulations are one-step cleaner/disinfectants registered with the US Environmental Protection Agency (EPA). Neutral or acidic cleaners are commonly used in cleanrooms for the purpose of periodic residue removal or to remove excessive soiling or particles after a worst-case event. After the application of a cleaner using a polyester or microfibre mopping routine with a double or triple bucket system, it would be best practice to employ a rinse step with Water for Injection (WFI).
Disinfection: The most common disinfectants utilised globally in biopharma and medical device cleanrooms are quaternary ammonium (quads) disinfectants and phenolic disinfectants. Quat disinfectants are known to be excellent cleaners with deodorising abilities. Phenolics are derived from coal tar (carbolic acid) and creosote. As an active ingredient, phenolics can have slightly higher levels of kill than quats against some microorganisms such as Mycobacterium.
The chemicals attack microorganisms in both broad and specific ways. For example, products with extremely low or high pH levels may present an inherently hostile environment to certain organisms, while the active ingredients (e.g. ortho phenyl phenol or benzalkonium chloride) may act on cell protoplasm or cell surfaces. In other words, not all disinfectants work the same way against all organisms. Further, not all organisms have the same susceptible cell structures. There are real differences in susceptibility between a vegetative bacterium and a bacterial spore. However, there also may be differences between the susceptibility of strains of the same species. Disinfectants should be selected on the basis of performance against common environmental isolates. Further, more than one product must be included in the disinfectant programme in order to obtain broad-spectrum performance. The spectrum should include routine disinfectants, sporicides, and alcohols.
Sporicidal agent: The disinfectant should be periodically rotated with a sporicide in an effort to address more resistant fungal and bacterial spores. The frequency of application of the sporicide would be based on environmental monitoring data and the frequency of isolation of spore-forming fungi and bacteria. Commonly used sporicides include sodium hypochlorite, hydrogen peroxide/peracetic acid blends, and hydrogen peroxide chemistries in liquid or vapour form.
Periodic rinsing may also take place to remove any disinfectant or sporicide residue build-up over time. WFI can be utilised with a damp mopping method using a two or three bucket system to remove residue built up on walls and floors quarterly, monthly, weekly, or bi-weekly based on visual observations. Detergents may also be considered as a means of removing disinfectant residue, for example for heavier residues on floor surfaces. Residue removal for glass and stainless steel can take place with a 70% isopropanol wiping step.
Disinfectant manufacturer’s testing and registration
Manufacturers in the US producing sanitisers, disinfectants, and sporicides are regulated by the







