The packaging of medical devices frequently takes place in a cleanroom, and the packaged devices normally undergo some kind of sterilisation. But it is the role of packaging to keep the device sterile right up until use. The correct selection of packaging, therefore, is a crucial but complex decision, and statistics suggest it is often not carried out correctly.
According to Jouni Vikman, Director for Healthcare at Wipak, 10% of medical device recalls are attributable to packaging failures and 31% of those are due to a hole in the packaging. Vikman chaired a recent Medical Device Packaging seminar in Oxford, UK. The seminar looked at the criteria for selection of packaging along with the regulatory requirements and testing procedures for the different types of packaging.
An overview of the current status of the relevant medical packaging standards was provided by Nina Tillaéus, a Member of the Board of the Sterile Barrier Association. Of the standards, ISO 11607-1 is among the most important. It states that medical device packaging should be:
- Made of known and traceable materials
- Non-toxic, non-leaching and odourless
- Free of holes, cracks, tears, creases and localised thinning
- Intended for use in medical applications.
It must also:
- Allow sterilisation
- Provide physical protection
- Maintain sterility up to the point of use
- Allow aseptic presentation.
Tillaéus said that when choosing the packaging materials and design, the key factors to consider are: the specific nature of the medical device and the intended sterilisation methods (e.g. ethylene oxide (EO), gamma irradiation, electron beam, steam, low-temp.oxidative); its intended use; expiry date; transport and storage.
| Medical Device Packaging standards |
| ISO 11607-1 Packaging for terminally sterilised medical devices, requirements for materials, sterile barrier systems and packaging systems |
| ISO 11607-2 Packaging for terminally sterilised medical devices, validation requirements for forming, sealing and assembly processes |
| ISO/TS 16775 – Guidance for use of ISO 11607 |
| EN-868 Parts 2-10 Packaging for terminally sterilised medical devices (wraps, pouches and reels…), particular requirements |
| ISO 13485 Medical devices – Quality management systems – Requirements for regulatory purposes |
ISO 11607-2 states that for ‘preformed sterile barrier system (SBS), SBS manufacturing processes shall be validated’. This would include, for example, processes such as rigid and flexible blister forming; pouch, reel or bag forming and sealing; form/fill/seal automated processes; kit assembly and wrapping; assembly of sterile fluid-path products; tray/lid sealing; filling and closing of reusable containers; and sterilisation sheets folding and wrapping.
EN-868 Parts 2-10 Packaging for terminally sterilised medical devices provides specific requirements for most kinds of medical packaging, including: sterilisation wraps, paper, paper bags, pouches and reels, paper for low temperature sterilisation, adhesive coated paper, containers, nonwoven (uncoated) and nonwoven (adhesive-coated).
Tillaéus highlighted recent updates on some of the existing standards. In May 2014, guidance on the application of ISO 11607 (ISO/TS 16775:2014) was published. Amendments were also published in July for ISO 11607-1 and -2. These include:
- Changes to definitions: 3.4 closure integrity, 3.8 microbial barrier, 3.19 seal integrity
- Adding ‘prevents the ingress of microorganisms, demonstrated under test conditions which consider sterilisation process, handling, distribution, transport and storage’
- An updated Annex, including new test methods (commercially available) and their status (see table B1 in the standard).
Tillaéus also flagged up potential future changes. For example, ISO/TC198/WG7 is carrying out a systematic review of ISO 11607-1 and -2, which is expected in early 2015, to which national standards bodies are required to respond by March 2015.
At the same time CEN/TC102/WG4 is looking at a revision of EN 868. This has already begun with the revision of EN868-2, -3, -4, -6 and -7. The other parts of EN 868 will be dealt with as soon as the above documents have been finalised. The next meeting is in March 2015 and publication is expected mid 2016, Tillaéus said.
Considerations for medical papers
Radka Peterson, Technical Sales for Medical Papers, BillerudKorsnäs, looked specifically at the ISO 11607 requirements for medical packaging papers. She highlighted the list of demands made on packaging materials, which include:
- to minimise safety hazards to both patient and user
- to protect the device during transport and storage
- to be non-toxic and be biocompatible
- to minimise the risk of contaminants to the patient and those involved in device transportation, storage and use
- to ensure that the device is sterile until point of use
- to be compatible with the sterilisation process
- to have an active life shelf-life of five years.
EN 868 parts 2-10 are referenced as informative documents in ISO 11607, she said, and while optional, they can be used to demonstrate compliance with parts of ISO 11607-1. They also provide detailed specifications for individual materials and packaging solutions. For example, for paper the characteristics listed in EN 868-3 and EN 868-6 include: grammage; air resistance; porosity; pore size; tear strength; wet and dry strength; tensile strength; burst strength; water absorption; water repellency; pH; fluorescent spots; and chloride and sulphate content.
The reasons behind the requirements are sometimes obvious, but not always. For example, pore size influences bacterial barrier performance; tear strength influences controlled tearing when opening; dry strength is for better protection of the contents during handling, while wet strength is required during steam and EO sterilisation.
Tensile strength is important for better protection of the contents and for speed when converting. Water absorption influences the bacterial barrier performance, as does water repellency. Chloride and sulphate content may affect the acidity of the material, which can affect coatings, inks and the medical device itself. Optical brightening agents are not allowed in medical packaging papers due to a possible toxic effect.
Peterson also looked at some of the particular properties required by the standards for: heat sealability and peel performance; clean peel performance; quick seal development; and an even and consistent seal.
Under the biocompatibility and toxicological requirement, which states that the material in contact with the device must not contain substances known to be toxic in sufficient quantity to cause a health hazard, Peterson highlighted that any possible breakdown materials from additives during the different sterilisation techniques must also be considered.
Under the requirement for cleanliness, visual appearance is a key element for a medical package, she said.
Some of the ‘enhanced requirements’ listed in the standards include: release characteristics – to provide a uniform seal; hold out – to provide a suitable ‘hold out’ for coatings and adhesives; and puncture resistance – indicating how a paper will perform if an object in the package impacts the paper.
Direct sealing of papers
Jonathan Andrews, Business Development Manager, Medical, BillerudKorsnäs, looked at the performance and characterisation of the direct sealing of papers. In the past, direct seals were weaker than seals formed using heat seal coated papers. Traditionally, the attempts to get the highest seal strength from a direct seal system resulted in unacceptably fibrous peels. However, with careful and continual product design he said this can now be avoided. The technology for direct seal paper has advanced significantly over the years and the objective assessment of the cleanliness of a peel as well as of seal strength is now possible, he said.
Not only do peeling ‘data libraries’ assist medical device manufacturers to optimise material combinations and sealing parameters, he said, but peels can also be engineered not to be sensitive to peel direction. Seal/peel strength and the cleanliness of the latest generation papers is now at a very high level, he added.
The advantages of direct seal include: reduced cost, maximised porosity and elimination of potential interactions between coatings and medical devices. Not using coatings does, however, require that the direct seal papers are of high quality and high performance.
Direct seal performance is governed by the properties of both sides of the pack, i.e. the paper top web and the film base web, or paper front web and film back web he explained. It also relies on the type of packaging/pouch making machine to be used, its set-up and condition, the direction of the peel with respect to the paper fibre lay, and the sterilisation processes.
Andrews looked at the minimum seal strength requirements detailed in EN 868-5:2009 Packaging for terminally sterilised medical devices – Part 5: Sealable pouches and reels of porous materials and plastic film construction – Requirements and test methods. This standard is cited in ISO 11607-1 as an approved test for seal strength. Para 4.5.1 of this standard states: ‘Minimum value for seal strength……..shall be 1.5 N/ 15mm for steam sterilisation processes and 1.2 N/15mm for other sterilisation processes’.
When assessing the quality of a direct seal a critical requirement (as per ISO 11607) is to ensure a sterile barrier has been achieved, he said. Seal assessment is split into two components:
- Assessments of seal strength – EN 868-5 Appendix D, ASTM F88 M-09 test methods are used to ensure compliance with ISO11607 (plus others)
- Cleanliness of peel – which depends on a visual, tactile and aural assessment.
The cleanliness has been measured subjectively until recently but more objective assessment techniques have now been developed. For example, peeled films samples are placed on a scanner and the scanned images are digitally compared with references to assess the level of fibre tear. The results are presented in the form of a numerical value between 0 and 4.
Seal strength and peel cleanliness data for a wide range of films and papers now form the basis for a data library that can be used to optimise paper and film combinations for a given packaging requirement. They can also be used to narrow down sealing parameters, thus saving time, resources and money during material selection and process validation.
To further characterise the direct sealing process, the use of advanced imaging is being investigated, said Anderson. The techniques being explored are scanning electron microscopy and thermography (see Figure 2).
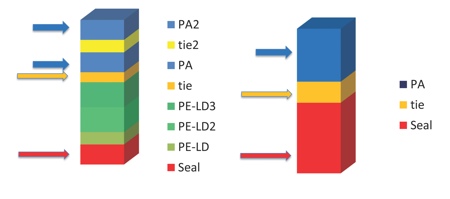
Figure 2: An example of multilayer formable film benefits, 8-layer multilayer film versus 3-layer film
Enables down-gauging, use of thinner structure for specified thermoforming ratio
Divided PA layers enhance thermoformability
Expensive individual layers can be thinner providing cost benefits
Several layers gives more security and wider choice of functional raw materials
Source: Wipak
Many factors influence the functionality of a direct sealing paper, including: fibre length, fibre refining, paper formation and sizing. Many paper properties depend upon the cellulose fibres from which the paper is manufactured; similarly the fibre length is directly related to the type of tree from which the pulp is manufactured. Typically, soft wood has long fibres and hard wood has short fibres. Long fibres enhance the physical strength of the papers and are much less likely to tear out of the paper on peeling.
‘Formation’ describes the alignment of the fibres in the paper. Unmodified, paper fibres tend to orientate themselves in the machine direction. This orientation results in a significant increase in the tendency for fibre tear at 180° to the fibre lay, and an increase at 90° to the fibre lay. This restricts the flexibility to align packs in the optimal orientation for a medical device manufacturer’s packaging process and leads to restrictions in print alignment, said Andrews.
Second-generation Direct Seal medical packaging papers are produced to ensure that the natural tendency for the fibres to align with the machine direction is eliminated, leading to a cleaner peel and more flexible product for the end user.
Finally, the paper surface can be modified with a very low weight coating (sizing) that does not have heat- or cold- seal properties. The size enhances seal strength and gives a very clean, omnidirectional peel. ‘Sized papers represent the best Direct Seal papers available today in terms of seal strength and clean peel,’ he said, adding that sizing can be applied to any weight of paper and to both standard and reinforced grades.
Medical films
Paavo Muhonen, R&D Manager, Wipak, looked at the manufacturing methods for medical films. He outlined the raw material requirements, the important processing parameters, and the quality considerations such as thickness control, in-line web camera control and cleanroom production. Under ISO 11607-1, the plastic raw materials in medical device packaging must meet the following:
- The source, history and traceability of all materials must be known
- Biocompatibility and toxicological attributes must be evaluated along with sterilisation effects on these according to ISO 10993
- Must not contain or release any toxic substances before, during and after sterilisation
- Must be FDA- and EU-compliant for food contact
- Must comply with the European Directive 94/62/EC on packaging and packaging waste, i.e. contain no heavy metals.
Film thickness is a key quality consideration, said Muhonen, and controlling it relies on inline measurement and gravimetric feeding systems – where because the feeding is based on the weight of material (i.e. density), it provides precise thickness control for each individual thin layer. In-line web cameras provide quality control and can detect defects smaller than 0.5mm. Other visual inspection systems and automatic marking systems can help manufacturers to pick up foreign particles such as gels, black particles and pinholes.
Cleanroom production also provides the required hygienic production environment and the requirements are covered in the Quality system ISO 9001, ISO 13485 for medical devices, and GMP (good manufacturing process), while bioburden limits are discussed in ISO 11737.
Multilayer films
When looking at medical grade film properties, Muhonen said the main considerations are: the packaging method, the cost and nature of the device; shape, size and weight of the device, and the sterilisation resistance. ISO 11607-1 requires that it be demonstrated that the materials are suitable for a specified sterilisation process and Muhonen suggested this is the first matter to check when starting to choose packaging materials. Common mistakes to avoid are the use of low melting point materials in steam sterilisation or using non-radiation resistant materials (such as standard polypropylene) in gamma or beta sterilisation.
Shelf life and stability testing are also required by ISO 11607-1. Section 6.4 deals with stability testing and says it should be performed using real time ageing protocol. It also says ‘Accelerated ageing shall be regarded as sufficient evidence for claimed expiry dated until real time ageing data is available.’
It should have a shelf-life of five years after the sterilisation process, and it should be noted that shelf life is not only a time-related issue but also event-related, explained Muhonen. For example, an incursion above a certain temperature or humidity or exposure to direct sunlight would affect shelf-life. This leads to the last point that storing conditions for the final packaging have to be correct in terms of humidity and temperature.
Muhonen went on to look at the growing trend for the use of multilayer films, which comprise functional layers that enable property tailoring, such as formability, mechanical strength, barrier protection for oxygen and water vapour.
According to Muhonen, it is all about ‘mastering the recipe such that the film fulfills the requirements of the customer’s final application’. An outer surface can be provided that protects the inner layers and can facilitate a wider sealing window. Alternatively, a polyamide (PA) layer can be added (outside or embedded) for improved forming and mechanical properties; a sealing layer allows fast sealing and peel performance; or a ‘bulk’ layer can be placed inside for better cost-efficiency.
Multilayers mean that thinner films can be used without compromising security and they enable the down-gauging and use of thinner structure for a specified thermoforming ratio. In addition, divided PA layers can enhance thermoformability and expensive individual layers can be made thinner giving further cost benefits. ‘Overall, several layers give more security and a wider choice of functional raw materials,’ he said.
Antibacterial additives
The addition of antimicrobial agents to packaging materials has been adopted by some industry sectors for a while and some believe that a step change in hygiene can be achieved through their use in medical packaging. Paul Morris, MD, Addmaster, a company offering permanent antibacterial protection for products, said there are currently more than 40 products with ‘Biomaster’ antibacterial protection being used in a medical setting ranging from floors, bed frames, uniforms and case note folders to vacuum sacks.
The efficacy of different antibacterial additive methods varies, but some companies have made progress in improving this. Morris explained that Biomaster binds to the bacteria cell wall, disrupting growth. The silver-complexed ions interfere with enzyme production, stopping the cell producing energy. It also interrupts the cell’s DNA, preventing replication. Wipak has a product approved to ISO 22196 that is suitable for medical devices and survives the sterilisation process, he said.
Chris Wright, Director of the Multidisciplinary Nanotechnology Centre, College of Engineering at Swansea University, also ran a workshop looking at future potential for antibacterial additives in medical packaging, particularly in view of the cost burden on health authorities of healthcare acquired infections and also in view of the move towards increasing home care and self administration of patients, where device use will no longer be carried out by trained professionals.
Stefan Krakow, Head of Product Management, GEA Tiromat Packaging, looked at process qualification for packaging machines. He discussed the requirements of machine validation and looked at the different stages involved and at assembly.
The main places to go for more information on this topic were: the US FDA’s Process Validation: General Principles and Practices, which offers general validation guidelines but which are open to individual interpretation, along with guidance from the International Society for Pharmaceutical Engineering (ISPE), and Good Automated Manufacturing Practice (GAMP) 5.
Fitness for purpose
Noel Gibbons, Lead Programme Manager, Anecto, looked at testing procedures for final medical device packages required by the standards. He reminded delegates of the five key purposes of device packaging: to contain, inform, protect, display and transport. He looked at the tests used to validate sterile integrity and strength of the primary package: Visual inspection, burst test, peel/tensile test, dye penetration, bubble leak, microbial challenge and gas trace (Sniffer) testing.
The Visual Inspection ASTM F1886 test, for example, covers the determination of channels in a package seal down to approx 75μm (0.003in.) with a 60–100% probability. This qualitative measure (accept/reject) helps to evaluate the appearance of unopened seals to determine the presence of defects. Key points to think about, he said, are: have you evaluated your sealing process during OQ and PQ? And have you set accept/reject levels for your sealing process?(see Figure 3).
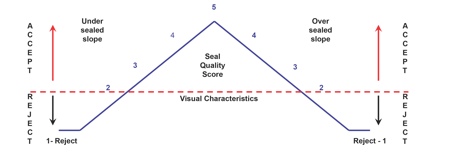
Figure 3: Visual inspection attribute qualification for medical device packaging
Visual Characteristic Rating Scale
5 – Acceptable: Package meets highest visual requirements
2 to 4 – Acceptable: Package meets seal requirements (under sealed or over sealed)
1 – Rejectable: Package seal does not meet minimum requirements
Source: Anecto
In his second presentation on package performance testing, Gibbons gave a comparison of different ASTM-ISTA criteria. He said many medical device manufacturers struggle with deciding what they need to do to comply with ISO 11607 and which procedure to utilise for performance testing – ASTM or ISTA. ASTM or ISTA standards are widely used and are listed in ISO 11607, a recognised consensus standard by the FDA, therefore either ASTM or ISTA test procedures can be employed, he said.
What this event highlighted, is that the time required to gather all this packaging data means that packaging requirements must be considered in parallel with the device development, if costly delays to market are to be avoided.






