First used in the 1940s, ethylene oxide (EO) is an alkaline gas that can infiltrate packaged medical devices and kill micro-organisms to achieve sterilisation. It is suited to the majority of heat-, moisture- or radiation-sensitive medical products. Because of EO’s reactive nature, the sterilisation process is carried out in enclosed chambers to prevent leaks and exposure to the gas.
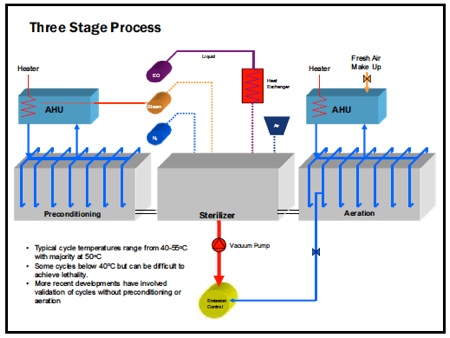
It is a slightly lengthier process than other methods, involving three stages:
- Preconditioning and/or conditioning of device through temperature and humidity variations
- The gas dwell phase/sterilising cycle where the device is exposed to the EO gas
- Aeration of the device for removal of gas from the product.
Featured companies








