
Manufacturing parenteral medicines requires a controlled and validated clean production environment. If the terminal sterilisation of the final product is not possible, aseptic manufacturing is the only alternative.
In aseptic production, exposure of the sterile product to the environment may take place during many different stages of the manufacturing process. Aseptic manufacturing of sterile products requires a high level of contamination control.
According to current Good Manufacturing Practices (GMP) guidelines, processes, equipment, facilities and manufacturing activities should be managed by applying Quality Risk Management (QRM) principles1 that provide a proactive means of identifying, scientifically evaluating and controlling potential risks to quality.
An important risk factor in sterile manufacturing is personnel. Contamination from people mainly consists of hair, skin cells, saliva, sebaceous matter, sweat, particles from clothing and exogenous particles and substances picked up in the environment. Adequate cleanroom garments, as well as undergarments, are critically important to minimise the risk of contaminating the environment or products.
Risk factors with cleanroom garments include gowning procedures and processes, as well as laundering, packing, sterilisation, repairs, storage, handling and logistics
Other risk factors with cleanroom garments include gowning procedures and processes, as well as laundering, packing, sterilisation, repairs, storage, handling and logistics.
Because many factors contribute to the overall quality and suitability of cleanroom garment systems, a risk- and science-based validation of cleanroom garment systems is very important. In this article, cleanroom garment systems are viewed as a process that will be validated in the frame of the overall aseptic process.
The term "validation" is used to aggregate the various qualification steps that are required to demonstrate that a cleanroom garment system is suitable for its intended use in a given aseptic process.
In this holistic innovative approach, aligned with quality-by-design principles2, more effort is spent at the front-end in the design phase and during design qualification. This will lead to designed-in risk reductions; better understanding of key aspects, limitations and residual risks; and fewer issues during final simulation runs and routine operations.
Regulatory guidance
Depending on the jurisdiction, aseptic production of sterile medicines must meet various regulatory requirements, such as those set out in: Annex 1 of the EU Guidelines to Good Manufacturing Practice; US Food and Drug Administration (FDA) Guidance for Industry on sterile drug production; or Japanese Guidance on the Manufacture of Sterile Pharmaceutical Products by Aseptic Processing.
Current EU-GMP guidelines require the use of sterilised or adequately sanitised garments for grade A/B areas and a written procedure for changing and washing that is designed to minimise contamination of clean area clothing or carry-through of contaminants to the clean areas. Reusable garments are required to be cleaned and handled in a way that the garment does not gather additional contaminants that can be shed later.
The current EU-GMP Annex 13 for the Manufacture of Sterile Medicinal Products includes little guidance on cleanroom garment qualification, except that it needs to be "appropriate".
Reusable garments should be replaced based at a set frequency determined by qualification
In contrast, the new draft EU-GMP Annex 14, published for consultation in December 2017, explicitly introduces the application of QRM principles and provides more details on gowning, including the requirement that gowning is part of a holistic contamination control strategy. This EU-GMP Annex 14 draft also requires that garments must be sterile and visually checked for cleanliness and integrity.
Another key addition is the requirement that "reusable garments should be replaced based at a set frequency determined by qualification or if the damage is identified." This compels manufacturers to produce data regarding the effect of reprocessing on the fabric and the overall garments. The ISO 14644-5: 2004 Annex B on cleanroom clothing requirements provides guidance that can be used to establish the user requirements specification (URS).
The ISO 13408-1: 2008 includes some general requirements on cleanroom garments for aseptic processing, but does not provide much guidance on cleanroom garment system qualifications.
IEST-RP-CC003.4: 2013 provides guidance on design, selection, specification, maintenance and testing of garment systems. Appendix B proposes tests for assessments of particle penetration and garment cleanliness. It is the most useful document to support the qualifications of cleanroom garment systems.
Validation approach
The four stages used for validation of equipment, facilities, utilities and systems can be applied to the validation of cleanroom garments. Some stages will focus on the quality of the cleanroom garment, but others must include additional components of the cleanroom garment system.
Packaging of the cleanroom garments should be part of the validation. Figure 1 provides an overview of the validation stages for cleanroom garments. Each validation stage must be formally finalised before progressing to the next stage.
First, a URS must be established that specifies the requirements necessary to create a feasible design that meets the intended purpose of the cleanroom garment. It may include additional requirements, such as the protection of people against chemical and/or biological agents.
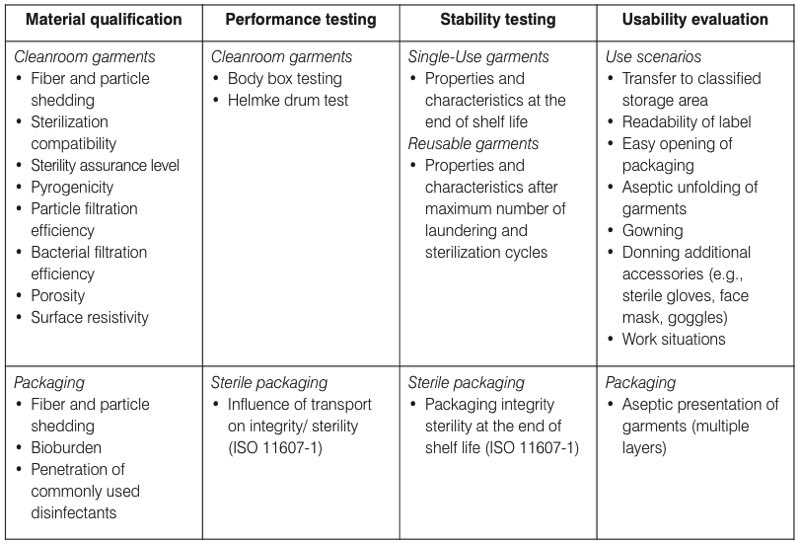
Table I. The four key areas of the design qualification (DQ) for cleanroom garments
used in EU-GMP grade A/B cleanrooms
Stage 1: Design qualification (DQ)
During DQ, compliance of the cleanroom garment design with cGMP must be demonstrated and documented, and the requirements of the URS must be verified to confirm that the selected cleanroom garment is qualified for the intended use.
The DQ must be executed and authorised by qualified, knowledgeable persons who can then challenge the proposed design and its performance to improve it.
Following the model of design validations of sterile barrier systems described in ISO 11607-1: 2019 - Packaging for terminally sterilised medical devices, it is recommended to split the DQ into four key areas. Relevant items for each of these areas are presented in Table I.
- Material qualification – includes the qualification of key characteristics and properties of the materials and fabrics used, the cleanroom garments and the packaging.
- Performance testing – includes testing the cleanroom garments and packaging under simulated and standardised conditions using standardised test methods.
- Stability testing – performed to assure that key material characteristics and properties remain sufficiently stable during the life cycle. Characteristics and properties that change over time should be validated under worst-case conditions.
Information for these three areas is normally provided by the supplier; however, it is important to verify that the data has been generated using validated and sound scientific methods.
- Usability evaluation – performed by the end-user to assure that the cleanroom garments can be used with acceptable remaining contamination and safety risks. Suppliers can also evaluate their garments for the intended use and supply that data to end-users for verification and further mitigation of identified risks during gowning and operations.
It is important to note that the validation of reusable cleanroom garments is a more complex process compared to single-use cleanroom garments. Repeated laundering, repeated sterilisation, multiple uses and repairs inherently influence the quality of reusable cleanroom garments.
The influence of these factors must be validated throughout the entire life cycle. In addition, not only the garment supplier must be qualified, but also the cleanroom laundry, sterilisation facilities and repair service. Reprocessing should be the subject of a separate DQ by the manufacturer and IQ-OQ-PQ by the supplier.
Stage 2: Installation qualification (IQ)
The IQ is a formal check to verify if all required elements are present, including facilities and Standard Operating Procedures (SOPs) for gowning and de-gowning; certificates of conformance and/or analysis; and the operator training and qualification plan.
Risk assessments executed as part of the DQ should be finalised and risk controls should be implemented. Figure 1 summarises the items to be included in the IQ.
Stage 3: Operational Qualification (OQ)
All relevant steps of the gowning and de-gowning process, as well as the aseptic presentation of the garments, should be qualified during the OQ (see Figure 1).
At least three independent, consecutive visual and microbiological assessments for at least one person who is trained for aseptic gowning. The OQ should also include a formal assessment, using all available sizes of the cleanroom garments and with people of lots of different body shapes. This verifies that all range of movements such as bending, stretching and lifting can be executed properly by any person that may need to use the garment.
Stage 4: Performance qualification (PQ)
The objective of the PQ is to validate the performance of the cleanroom garment system in use. The PQ is typically performed under worst-case conditions, which are determined based on a risk assessment.
Prior to performing the PQ, it is necessary to define the actions that should be taken if established criteria are not met. During the first phase, compliance with aseptic gowning procedures should be confirmed using both a visual and microbiological assessment.
Each person accessing an EU-GMP grade A/B environment must perform a gowning qualification. Only adequately trained persons should execute the PQ to exclude failures due to causes other than quality issues with the cleanroom garments.
The second phase focuses on the validation of the microbiological quality of the gowned personnel while performing work tasks (e.g. aseptic compounding, cleaning and disinfecting). This phase also includes validation of the microbiological and particulate quality of the work environment and the execution of aseptic process validations.
Revalidation and change management
To confirm that the cleanroom garment system remains in a state of control, it should be evaluated at an appropriate frequency (e.g. annually or biennially). Gowning qualifications should be repeated at least once a year, or more frequently if there is any doubt about the quality of the gowning process or skills of specific persons.
Changes must be reviewed critically and may lead to revalidations. Well-documented DQs, as well as IQ-OQ-PQ, are the basis for successful change management. A science- and risk-based, and quality-by-design approach offers many benefits when applied to the design, selection and implementation of cleanroom garments.
It is not only the correct approach to effectively control contamination risks related to people, but it is also an appropriate response to the latest regulatory requirements, such as the new EU-GMP Annex 1 draft that is based on QRM principles and introduces the concept of a holistic contamination control strategy (CCS). The CCS considers all aspects of contamination control during the entire life cycle, based on thorough technical knowledge and sound process know-how. This approach also creates a foundation for proper root cause analysis in case of deviations, and for risk-based change management.
References
- International Council for Harmonisation (ICH). ICH Q9 - Quality risk management. In:2005
- International Council for Harmonisation (ICH). PHARMACEUTICAL DEVELOPMENT Q8(R2). In: INTERNATIONAL CONFERENCE ON HARMONISATION OF TECHNICAL REQUIREMENTS FOR REGISTRATION OF PHARMACEUTICALS FOR HUMAN USE, ed2009
- European Medicine Agency (EMA). Annex 1 - Manufacture of Sterile Medicinal Products - draft for public consulation. In: European Medicine Agency (EMA), ed. Brussels: Directorate-General for Health and Food Safety, Unit SANTE B/4, BE-1049 Brussels; 2017
N.B. This article is an edited version of the technical paper originally published in the Journal of GXP Compliance, Volume 23, Issue 4, Jul 2019, and featured in the November 2019 issue of Cleanroom Technology.
Subscribe today and get your print copy! The latest digital edition is available online.



