Not only has the length of Annex 1 been increased from 16 to 50 pages, but the whole approach has changed, which will have repercussions on the technologies and the procedures used in pharmaceutical manufacturing.
It anticipates that all pharmaceutical manufacturing activities will be governed holistically by the QRM principles and documented in the contamination control strategy (CCS). The CCS will become a living document, based on a data-driven scientific approach, which should be continuously updated and improved in order to control potential risks to quality.
The new draft is calling for a proactive approach: simply reacting to and correcting detected contamination will no longer be enough. Manufacturers will be expected to fully understand their processes and procedures, so that they can identify the potential risks to quality, put in place all the technical and procedural means to control these risks and aim for continuous improvements.
Since cleanroom garment systems are a critical part of sterile and aseptic manufacturing, they need to be managed under QRM (Quality Risk Management) principles too.
QRM principles for cleanroom garments
Quality risk management starts with an analysis and understanding of all the risks to quality associated with cleanroom operators wearing cleanroom garments. A complete data-based analysis will allow for design certification, qualification, validation and monitoring procedures which have quality built into them, thus being part of a holistic contamination control strategy.
A risk analysis is needed to understand the contamination risks coming from operators wearing cleanroom garments. Operators represent the biggest source of contamination inside cleanrooms, responsible for 75% of all contaminants. This contamination is coming both from the operators themselves and from their cleanroom garments.
The EU guidance advises on a validation approach that consists of five steps
Operator contamination is due both to our human nature (an average person sheds 40 000 particles per minute and 10% of them carry microorganisms) and human behaviour. While it is possible to mitigate the latter aspect through careful operator selection, training, slow movements or impeccable hygiene, the fact is that operators will always be shedding particles, as multiple studies have proven.
There is just one way to prevent particles generated by operators from contaminating the cleanroom: use cleanroom garments. They are the only barrier between the operator and the production environment.
The 2020 draft of Annex 1 clearly points this out: “(the cleanroom garments should) retain particulates shed by the body”.
Cleanroom garments themselves may be a source of contamination and this risk needs to be assessed too.
For example, the material used for making the garments (non-woven for single-use garments or woven for reusables) can shed more or fewer particles depending on the nature of the fibres or filaments used, their resistance to abrasion or their construction. The trims (zipper, buttons, elastic or sewing threads) may also be a source of contamination.
The design of the garment plays a role too and should be evaluated. One detail which is often neglected is the packaging in which the cleanroom garments come, which could be an additional source of contamination (i.e. paper-back bag vs. plastic bags).
Main stages of validation
Once the risks have been evaluated, they should be removed or replaced by technical or organisational means as far as possible, and the residual risks mitigated as much as possible using a validated cleanroom garment system.
The EU general guidance on validation (GMP Annex 1519) provides the general framework which can be applied to the qualification of cleanroom garment systems as well. This validation approach consists of five steps: the definition of User Requirements Specification (URS), the Design Qualification (DQ), the Installation Qualification (IQ), the Operational Qualification (OQ) and the Performance Qualification (PQ).
While the DQ and IQ have the highest impact on the quality achieved, the other stages should not be neglected, and it is important to proceed step by step.
User Requirements Specification (URS)
While they are not formally part of the validation process, it is important to define upfront the requirements on the cleanroom garments system from the users and the environment they work in.
The URS will define the critical requirements against which the garment system needs to be assessed so that they will be in line with the risk assessment.
For example, a trained operator may have to be able to work at least 3 hours in the same set of cleanroom garments without causing unacceptable (cGMP) levels of contamination of the garments and the aseptic working environment. The garment’s packaging system may have to be suitable for the layout of the cleanroom and its material pass-through systems, or may have to be suitable for manual spray disinfection.
Sometimes, the operator may also need chemical or biological protection against the substances they are handling inside.
Design Qualification (DQ)
The compliance of the cleanroom garment system with cGMP must be demonstrated and documented during the DQ, which aims to confirm that the selected cleanroom garment is qualified for the intended use.
As the new Annex 1 will require a data-driven scientific approach, the DQ should include tests to simulate the intended use and the performances of the garments. As recommended by ISO 11607-1, the DQ should be split into four key areas: Material qualification, Performance testing, Stability testing and Usability evaluation. For reusable garments this needs to be extended to the garment maker’s subcontractors, suppliers and service providers.
M. Pavičić and T. Wagner have listed properties which should be assessed in table 1 below.
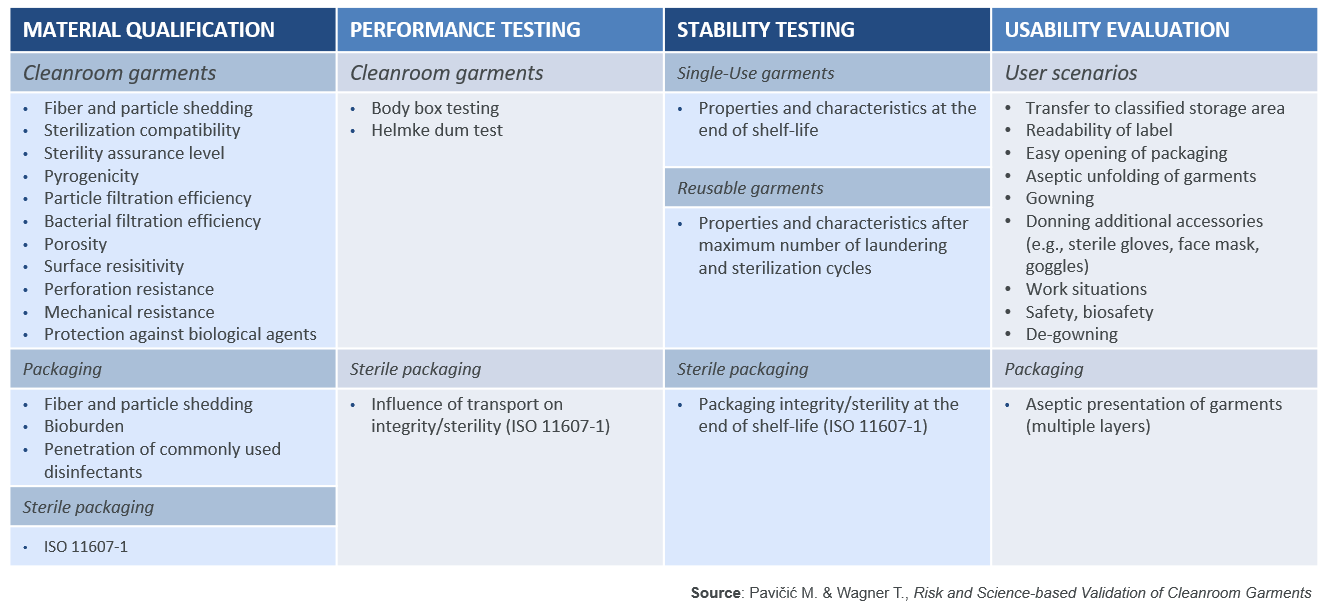
In this article, only a couple of these properties will be highlighted to show their importance and the scientific test methods which may be used to assess the performance of cleanroom garment systems.
1. Material qualification: In order to ascertain whether the garments are truly sterile, it is important to check if the manufacturer is following a validated sterilisation process and can guarantee a sterility assurance level of 10-6 as per ANSI/AAMI/ISO 11137-1 and document this in a certificate of sterility. A simple certificate of irradiation or a document attesting an internal autoclaving process are not enough.
Since the cleanroom garments need to be a barrier against the human contamination generated by the operators, it is important to assess the filtration efficiencies of the materials (non-woven or reusable polyester fabrics) used for making the garments. The particle filtration efficiency (PFE) against dry particles can be assessed with the test method EN 143 (TSI 8130), which measures the filtration efficiency using salt particles having a diameter of 0.3µm. While the bacterial filtration efficiency (BFE) can be assessed with the test method ASTM F2101.
2. Performance testing: The Helmke Drum test method as per IEST-RP – C003.4 is a good way to assess the particle shedding of cleanroom garments, especially for garments that are washed multiple times.
The Body box test (IEST-RP-CC003.4) is the only test method available to assess particle shedding when a garment is being worn by an operator. It allows evaluation of both the particle shedding of the garment and its PFE & BFE of the particles shed by the operator.
3. Stability testing: It is important to check how the garment characteristics and properties will change over time (due to ageing, wear, and wash-dry-sterilisation cycles). Therefore the performances listed above should be validated under worst-case conditions, i.e. for single-use garments, assessing garments from different batches and at the end of their shelf-life, and for reusable garments after 10, 20, 30, 40 and 50 wash-dry-sterilisation cycles to assess the end-of-life of the garments (studies by Romano F., Ljungqvist B. and B. Reinmüller have demonstrated that repeated washing deteriorates garment performance).

Usability evaluation
It is important to go through the user scenarios and to assess the packaging of the garments to ensure that the cleanroom garments can be used with acceptable remaining contamination and safety risks. While this is typically done by the end-user, suppliers can also evaluate and supply data to users.
Installation Qualification (IQ)
Even though the IQ is a formal check to verify if all required elements of the cleanroom gowning system are present, it is important to check the following in order to eliminate unforeseen risks: Are the gowning and de-gowning facilities in order? Did the supplier provide the required certificates of conformance and/or analysis, supplier instructions, etc? Have the SOPs for gowning and de-gowning been written or adapted? Have the logistical processes for garments and accessories been validated? Has the operators’ training and qualification plans been established?
Operational Qualification (OQ)
The OQ aims to qualify the gowning and de-gowning concept, including logistics & material pass-throughs, and the aseptic presentation of the garments (i.e. folding, packaging).
Performance Qualification (PQ)
The PQ is typically done under worst-case conditions, which must be determined based on a risk assessment to validate the performance of the cleanroom garment system when it is used. The requirements specified in the URS must be complied with fully. They include the aseptic gowning qualification and the validation of the microbiological quality of the gowned personnel with the garments and other accessories during the actual work.
PQ is typically done under worst-case conditions
Of course, it does not end there: periodic revalidation of the garment system, constant monitoring and critical review of changes to the garments or the system are important to demonstrate the state of control.
Cleanroom garment systems are a critical part of the contamination control strategy and process validation. A Risk and Science-based Quality-by-Design approach and verification is the correct strategy to control contamination risks related to people and offer designed-in risk reductions. This approach is an adequate response to the latest regulatory requirements.
